 2020, Vol. 31
2020, Vol. 31 


b Department of Chemistry, Tsinghua University, Beijing 100084, China;
c Department of Chemistry, University of Science and Technology of China, Hefei 230601, China;
d Bayer AG, Department of Medicinal Chemistry, Aprather Weg 18 A, 42096, Wuppertal, Germany
The anaphylatoxin C5a, a proinflammartory mediator containing three pairs of disulfide bonds, is a highly potent endogenous activator of chemoattractant receptor C5aR [1, 2] involving in the inflammatory process of tissue damage. C5a-associated excessive response may cause or aggravate a variety of inflammatory and autoimmune diseases, such as rheumatoid arthritis, neuropathic and inflammatory pain, Alzheimer's disease and cancer [3-6]. Therefore, the development of antagonists of C5a has been of high interest to the pharmaceutical researches [7, 8]. Although a variety of active molecules that mimic C5a have recently been reported, such as PMX-53, they still have limitations, including insufficient antagonistic activity and in vivo short half-life, which have hampered their application in clinical trials [9, 10]. To overcome these limitations, there is a strong need to gain an in-depth understanding of the structure-activity relationship of the natural C5a ligand for the goal of the development of next generation of C5a antagonists.
Previous studies have shown that C5a exhibits its biological function primarily through its C-terminal polypeptide sequence which interacts with the hydrophobic pocket of the transmembrane domain of C5aR [11, 12]. An important and interesting phenomenon is that the last amino acid at the C-terminus of C5a, that is Arg, has been found to play a key role in controlling the in vivo activity of C5a. Removal of the C-terminal Arg residue of C5a in plasma by the carboxypeptidase results in a 1000-fold reduction in allergic reactions [13, 14]. In addition to the fact that this Arg may be directly involved in the C5a-C5aR interaction, another important factor leading to this phenomenon is that Arg at the C5a C-terminus may also regulate its C-terminal conformation. The structures of human C5a and human C5a-desArg (desArg denotes that the C-terminal arginine is deleted) have recently been reported. Unfortunately, the C-terminal eight residues of previously reported crystal structure of human C5a-desArg are not visible, indicating structural instability of this peptide sequence under crystallization conditions [15, 16].
Herein, we reported the total chemical synthesis of rat C5adesArg and its D-enantiomer, namely L-rC5a-desArg and D-rC5adesArg, and for the first time obtained the crystal structure of rC5adesArg by using racemic crystallography. Rat C5a (rC5a) and human C5a (hC5a) are highly conserved in sequence, and thus the rC5a-desArg structure could shed light on how the C-terminal Arg of human C5a regulates its C-terminal conformation. Our rC5adesArg structure allows us to clearly analyze the C-terminal structure of rC5a-desArg. Unlike the previously reported human C5a structure, the C-terminal sequence of rC5a-desArg extends from H4 in a continuous helical structure, which gives rC5a-desArg a longer H4. This result indicates that Arg does affect the Cterminal conformation of C5a, which may have an effect on the biological activity of C5a and may help us to optimize the C5a antagonists.
Our study began with the chemical synthesis of full-length rC5a-desArg. The chemical synthesis method can obtain D-C5adesArg, which is a prerequisite for the racemic crystallography and is inaccessible by the recombinant protein expression [15]. The racemic crystallization technique is capable of obtaining high quality crystals of rC5a-desArg that are not obtainable by chiral pure L-rC5a-desArg [17-19]. On the other hand, we obtained the crystal structure of human C5a-desArg by racemic crystallography, however, its C-terminal eight residues of human C5a-desArg are also not visible. Our study results indicate that only rat C5a-desArg could provide high quality crystals for X-ray diffraction. Therefore, though both human and rat C5a-desArg were made during our study, we only described the chemical synthesis of rC5a-desArg.
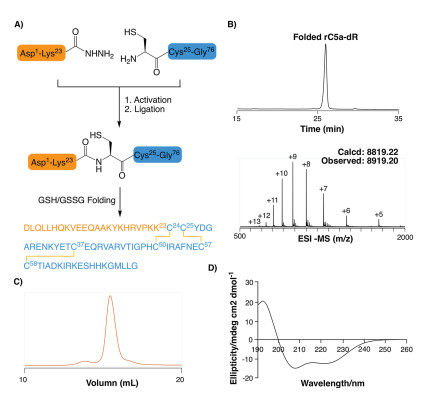
The chemical synthesis of rC5a-desArg was carried out by Fmoc-based solid phase peptide synthesis (SPPS) [20-29] and hydrazide-based native chemical ligation [30-35]. Specifically, Cys24 was selected as a ligation site to divide rC5a-desArg into two polypeptide fragments, namely, H-[Asp1-Lys23]-NHNH2 and H- [Cys24-Gly76]-OH (Fig. 1A). These two fragments were prepared from hydrazine-Trt(2-Cl) and Wang resins with isolated yields of 54% and 46%, respectively.

|
Download:
|
| Fig. 1. Synthesis and characterization of rC5a-desArg. (A) Synthetic route of rC5adesArg; (B) Analytical RP-HPLC chromatogram (λ = 214 nm) and ESI-MS analysis (observed mass =8819.20 Da, calculated mass = 8812.22 Da) of folded rC5a-desArg; (C) Gel filtration chromatography of folded rC5a-desArg (Superdex 75 column (10/ 300 G L), λ = 280 nm); (D) Circular dichroism spectrum (190-260 nm) of folded rC5a-desArg. | |
{kind=link}
After sequential NaNO2 oxidation and treatment with 4- mercaptophenylacetic acid (MPAA), H-[Asp1-Lys23]-NHNH2 was efficiently converted to the thioester intermediate H-[Asp1-Lys23]-MPAA. This thioester peptide was subsequently ligated with H-[Cys24-Gly76]-OH by native chemical ligation to afford full length rC5a-desArg in an isolated yield of 52%. After purification by reverse phase high performance liquid chromatography (RPHPLC), the ligation product rC5a-desArg was subjected to in vitro folding at pH 7.4 under conditions containing a reduced/oxidized glutathione pair [36, 37]. The folding process reached equilibrium in 6 hours at room temperature with an isolated yield of 68% (Fig. 1B), as confirmed by RP-HPLC and electrospray ionization mass spectrometry (ESI-MS). Subsequently, the well-folded rC5adesArg was further purified by size exclusion chromatography (SEC). SEC results showed that the folded rC5a-desArg was eluted as a single peak (Fig. 1C). Circular dichroism (CD) spectroscopy analysis of the folded rC5a-desArg revealed characteristic peaks at 222 and 208 nm, similar to that of the previously reported recombinant porcine C5a (Fig. 1D) [38].
With the folded rC5a-desArg in hand, we carried out the crystallization experiments. Unfortunately, no good-quality crystals were obtained after repeated trials. Previous studies have shown that crystals could grow more readily in racemic mixtures than with pure chiral molecules because of the additional achiral space groups provided by racemic mixtures [39-45]. Consequently the technology of racemic crystallization has recently been used in increasing cases to obtain the crystal structure of the difficult-tocrystallize proteins [46-49], such as snow flea antifreeze protein (sfAFP) [50] and potassium channel blocker protein BmBKTx1 [51]. Here, we attempted to elucidate the structure of rC5a-desArg by using racemic X-ray diffraction crystallization.
For racemic crystallization, we initiated to synthesize D-rC5adesArg. The synthetic route for D-rC5a-desArg is shown in Fig. 2A. The polypeptide fragments H-[(D)Asp1-(D)Lys23]-NHNH2 and H- [(D)Cys24-Gly76]-OH were prepared by standard Fmoc SPPS under microwave conditions, and the isolated yields were 71% and 32%, respectively. The hydrazide-based native chemical ligation and subsequent oxidative folding provided a folded product of molecular weight 8801.00 Da, indicating that the three pairs of disulfide bonds were successfully formed (Fig. 2B). The SEC result showed good homogeneity of the folded D-rC5a-desArg (Fig. 2C). Furthermore, the CD spectrum of the folded D-rC5a-desArg was identical in shape to that of rC5a-desArg but the signal was reversed (Fig. 2D).

|
Download:
|
| Fig. 2. Synthetic route and characteristics of D-rC5a-desArg. (A) Synthetic route of D-rC5a-desArg; (B) analytical RP-HPLC chromatography (λ = 214 nm) and ESI-MS analysis of folded D-rC5a-desArg (observed mass =8801.00 Da, calculated mass =8801.00 Da, (D)met was mutated to (D)nle to avoid oxidation); (C) Gel filtration chromatographic (Superdex 75 colimn (10/300 G L), λ = 280 nm) of folded D-rC5a-desArg; (D) Circular dichroism spectrum of the folded D-rC5a-desArg (The circular dichroism spectrum of L-rC5a-desArg is indicated by the dotted line; (E) rC5a-desArg crystals formed by racemic crystallization (0.2 mol/L hydrogen phosphate diamine, 20% w/v polyethylene glycol) 3350 and pH 8.0. | |
{kind=link}
A racemic mixed solution containing equal amounts of L-rC5adesArg and D-rC5a-desArg was screened using 96 commercially available crystallization screening conditions to obtain crystals. Good-quality crystals were observed in about 10 conditions by the sitting vapor diffusion method at 18 ℃. This observation was in sharp contrast to the poor crystal forms when we tried to obtain crystals using L-rC5a-desArg alone. The best diffraction quality rC5a-desArg crystals were formed in 0.2 mol/L hydrogen phosphate diamine, 20% (w/v) polyethylene glycol 3350 and pH 8.0. The crystal form was quadrilateral (Fig. 2E). The diffraction intensity statistics showed that the space group of the crystal was P1 and the diffraction resolution was 1.80 Å. The rC5a-desArg structure was resolved by molecular replacement and refined to the final X-ray data statistics [52].
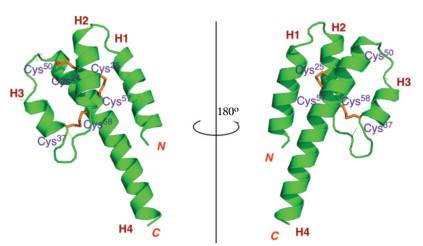
The X-ray structure of rC5a-desArg was shown in Fig. 3. In line with our expectations, the core structure of rC5a-desArg is an antiparallel four helices bundle stabilized by three pairs of disulfide bonds (Cys24-Cys50, Cys25-Cys57 and Cys37-Cys58), similar to the monomer A in the hC5a-desArg structure [15]. The monomer A also contained four helical bundles and three pairs of disulfide bonds (Cys21-Cys47, Cys22-Cys54 and Cys34-Cys55). The rC5a-desArg monomer contains four distinct helical structures, the N-terminal helix H1 consisting of the residues Asp1-Lys15, the second helix H2 containing the residues Arg19-Ala29, and the third helix H3 formed by the residues Cys37-Arg43 And the C-terminal helix H4 containing the residue Pro48-Gly76.

|
Download:
|
| Fig. 3. A cartoon representation of the structure of rC5a-desArg (green), with three pairs of disulfide bond (orange). | |
{kind=link}
Nuclear magnetic resonance (NMR) was previously used to determine the three-dimensional structure of hC5a, and the results showed that the core region of hC5a mainly consisted of four antiparallel-helix structures stabilized by three pairs of disulfide bonds. However, the structure of the C-terminal residues of human C5a remained unsolved. Zuiderweg et al. reported that the C-terminus of hC5a was disordered or adopted multiple conformations [53]. In contrast, Zhang et al. reported that the Met69-Arg74 sequence of hC5a was packaged back into the core region in a 1.5- turn helical structure and connected to the core region via a short loop (Fig. 4A) [16].

|
Download:
|
| Fig. 4. (A) Crystal structure of hC5a (1KJS), with the core region (blue-green), Cterminal structure (lemon); (B) The alignment of rC5a-desArg (the core region marked with green and the C-terminal structure marked with magenta) and hC5a; (C) Surface representation of rC5a-desArg and hC5a. | |
{kind=link}
The superposition of the structures of rC5a-desArg and hC5a showed that the core region of rC5a-desArg is similar to that of hC5a (Fig. 4B). In particular, the first, second and fourth helixes of C5a are almost completely superimposable. The main difference occurs at the third helix and C-terminus of C5a. The orientation of the third helix of hC5a is almost perpendicular to the fourth helix, whereas the orientation of the third helix of rC5a-desArg is almost parallel to the fourth helix. Importantly, the ten residues at the C-terminus of rC5a-desArg were visible in our structure. Previous studies have shown that the residues at the C-terminus of hC5a were packaged back into the core region, so that the C-terminal arginine residue approached Arg62 in space (Fig. 4C) [16]. These two adjacent Arg residues formed a positively charged surface to facilitate the binding of C5a to the receptor C5aR. In addition, the point mutation assay also showed that the mutations of C-terminal Arg and Arg62 did result in a decrease in the binding affinity to C5aR and agonist potency of hC5a. In contrast, the last ten residues of rC5a-desArg extend from H4 in a continuous helical configuration, which caused rC5a-desArg to have a longer H4. This significant conformational difference may indicate that the Cterminus of C5a is unable to form the positively charged surface when the C-terminal arginine is deleted, thereby reducing the receptor binding affinity and agonist activity of C5a.
In summary, we reported a robust method to prepare L-rC5a-desArg and its enantiomers by Fmoc-based SPPS and hydrazide-based native chemical ligation. Subsequently, using racemic protein crystallization, we obtained the X-ray structure of rC5a-desArg for the first time. Importantly, we found that the residues at the C-terminus of rC5a-desArg extended from H4 in a continuous spiral structure, which is significantly different from the structure of the C-terminus of hC5a. This structural change may explain the phenomenon that C-terminal arginine deletion results in distinct C5a biological activity and receptor binding affinity. These findings may also provide useful insights into the design of next generation of C5a antagonists.
AcknowledgmentsThe work was supported by the National Key R & D Program of China (No. 2017YFA0505200), the National Natural Science Foundation of China (Nos. 21532004, 21807001, 91753205, 81621002, 21621003).
Appendix A. Supplementary dataSupplementary material related to this article can be found, inthe online version, at doi:https://doi.org/10.1016/j.cclet.2019.08.039.
| [1] |
H. Liu, H.R. Kim, R.N.V.K. Deepak, et al., Nat. Struct. Mol. Biol. 25 (2018) 472-481. DOI:10.1038/s41594-018-0067-z |
| [2] |
A. Klos, E. Wende, K.J. Wareham, et al., Pharmacol. Rev. 65 (2013) 500-543. DOI:10.1124/pr.111.005223 |
| [3] |
M.L. Fonseca, R.R. Ager, S.H. Chu, et al., J. Immunol. 183 (2009) 1375-1383. DOI:10.4049/jimmunol.0901005 |
| [4] |
K.S. Nandakumar, A. Jansson, B. Xu, et al., PloS One 5 (2010) e13511. DOI:10.1371/journal.pone.0013511 |
| [5] |
E. Ting, A.T. Guerrero, T.M. Cunha, et al., Br. J. Pharmacol. 153 (2008) 1043-1053. |
| [6] |
A.S. Yekkirala, D.P. Roberson, B.P. Bean, et al., Br. Nat. Rev. Drug Discov. 16 (2017) 545-564. DOI:10.1038/nrd.2017.87 |
| [7] |
R.F. Guo, P.A. Ward, Annu. Rev. Immunol. 23 (2005) 821-852. DOI:10.1146/annurev.immunol.23.021704.115835 |
| [8] |
S.J. Bradley, A.B. Tobin, Annu. Rev. Pharmacol. Toxicol. 56 (2016) 535-559. DOI:10.1146/annurev-pharmtox-011613-140012 |
| [9] |
A. Moriconi, T.M. Cunha, G.R. Souza, Proc. Natl. Acad. Sci. U. S. A. 111 (2014) 16937-16942. DOI:10.1073/pnas.1417365111 |
| [10] |
D. Ricklin, J.D. Lambris, Nat. Biotrchnol. 25 (2007) 1265-1275. DOI:10.1038/nbt1342 |
| [11] |
M.S. Huber-Lang, J.V. Sarma, S.R. McGuire, J. Immunol. 170 (2003) 6115-6124. DOI:10.4049/jimmunol.170.12.6115 |
| [12] |
P.A. Ward, Nat. Rev. Immunol. 4 (2004) 133-142. DOI:10.1038/nri1269 |
| [13] |
Y. Laumonnier, A.V. Wiese, J. Figge, et al., Mol. Immunol. 84 (2017) 51-56. DOI:10.1016/j.molimm.2016.11.013 |
| [14] |
M.F. Li, Y.H. Hu, Dev. Comp. Immunol. 60 (2016) 139-148. DOI:10.1016/j.dci.2016.02.028 |
| [15] |
W.J. Cook, N. Galakatos, W.C. Boyar, et al., Acta. Cryst. D 66 (2010) 190-197. DOI:10.1107/S0907444909049051 |
| [16] |
X. Zhang, W. Boyar, M.J. Toth, et al., Proteins 28 (1997) 261-267. DOI:10.1002/(SICI)1097-0134(199706)28:2<261::AID-PROT13>3.0.CO;2-G |
| [17] |
S.B. Kent, Chem. Soc. Rev. 38 (2009) 338-351. DOI:10.1039/B700141J |
| [18] |
B.L. Pentelute, K. Mandal, Z.P. Gates, et al., Chem. Commun. 46 (2010) 8174-8176. DOI:10.1039/c0cc03148h |
| [19] |
Z. Wang, W. Xu, L. Liu, T.F. Zhu, Nat. Chem. 8 (2016) 698-704. DOI:10.1038/nchem.2517 |
| [20] |
J.B. Blanco-Canosa, P.E. Dawson, Angew. Chem. Int. Ed. 47 (2008) 6851-6855. DOI:10.1002/anie.200705471 |
| [21] |
B.L. Nilsson, M.B. Soellner, R.T. Raines, Annu. Rev. Biophys. Biomol. Struct. 34 (2005) 91-118. DOI:10.1146/annurev.biophys.34.040204.144700 |
| [22] |
W.H. So, C.T. Wong, J. Xia, Chin. Chem. Lett. 29 (2018) 1058-1062. DOI:10.1016/j.cclet.2018.05.015 |
| [23] |
X. Li, Y. Zhou, H.G. Hu, Chin. Chem. Lett. 29 (2018) 1088-1092. DOI:10.1016/j.cclet.2018.01.018 |
| [24] |
S. Tang, L. Liang, Y. Si, et al., Angew. Chem. Int. Ed. 56 (2017) 13333-13337. DOI:10.1002/anie.201708067 |
| [25] |
J. Zheng, M. Yu, Y. Qi, et al., J. Am. Chem. Soc. 136 (2014) 3695-3704. DOI:10.1021/ja500222u |
| [26] |
S. Tang, Y.Y. Si, Z.P. Wang, et al., Angew. Chem. Int. Ed. 54 (2015) 5713-5717. DOI:10.1002/anie.201500051 |
| [27] |
Y. Li, Y. Li, M. Pan, et al., Angew. Chem. Int. Ed. 53 (2014) 2198-2202. DOI:10.1002/anie.201310010 |
| [28] |
K. Jin, T. Li, H.Y. Chow, H. Liu, X. Li, Angew. Chem. Int. Ed. 56 (2017) 14607-14611. DOI:10.1002/anie.201709097 |
| [29] |
J. Yang, C. Wang, S. Xu, J. Zhao, Angew. Chem. Int. Ed. 58 (2019) 1382-1386. DOI:10.1002/anie.201811586 |
| [30] |
X.Q. Guo, J. Liang, Y. Li, et al., Chin. Chem. Lett. 29 (2017) 2744-2748. |
| [31] |
J.R. Liu, S.W. Dong, Chin. Chem. Lett. 29 (2018) 1131-1134. DOI:10.1016/j.cclet.2018.05.014 |
| [32] |
G.M. Fang, Y.M. Li, F. Shen, et al., Angew. Chem. Int. Ed. 50 (2011) 7645-7649. DOI:10.1002/anie.201100996 |
| [33] |
J.S. Zheng, S. Tang, Y.K. Qi, et al., Nat. Protoc. 8 (2013) 2483-2495. DOI:10.1038/nprot.2013.152 |
| [34] |
J. Li, Y. Li, Q. He, et al., Org. Biomol. Chem. 12 (2014) 5435-5441. DOI:10.1039/C4OB00715H |
| [35] |
G.M. Fang, J.X. Wang, L. Liu, Angew. Chem. Int. Ed. 51 (2012) 10347-10350. DOI:10.1002/anie.201203843 |
| [36] |
G.M. Fang, X.X. Chen, Q.Q. Yang, et al., Chin. Chem. Lett. 29 (2018) 1033-1042. DOI:10.1016/j.cclet.2018.02.002 |
| [37] |
Y. Ji, S. Majumder, M. Millard, J. Am. Chem. Soc. 135 (2013) 11623-11633. DOI:10.1021/ja405108p |
| [38] |
W.T. Morgan, E.H. Vallota, H.J. Müller-Eberhard, Biochem. Biophys. Res. Commun. 57 (1974) 572-577. DOI:10.1016/0006-291X(74)90584-1 |
| [39] |
K. Mandal, B.L. Pentelute, V. Tereshko, et al., Protein Sci. 18 (2009) 1146-1154. DOI:10.1002/pro.127 |
| [40] |
S. Gao, M. Pan, Y. Zheng, et al., J. Am. Chem. Soc. 138 (2016) 14497-14502. DOI:10.1021/jacs.6b09545 |
| [41] |
B.J. Yan, L.Z. Ye, W.L. Xu, et al., Bioorg. Med. Chem. 25 (2017) 4973-4965. |
| [42] |
T.O. Yeates, S.B.H. Kent, Annu. Rev. Biophys. 41 (2012) 41-61. DOI:10.1146/annurev-biophys-050511-102333 |
| [43] |
H. Yeung, C.J. Squire, Y. Yosaatmadja, et al., Angew. Chem. Int. Ed. 55 (2016) 7930-7933. DOI:10.1002/anie.201602719 |
| [44] |
S. Gao, M. Pan, Y. Zheng, et al., J. Am. Chem. Soc. 138 (2016) 14497-14502. DOI:10.1021/jacs.6b09545 |
| [45] |
M. Pan, S. Gao, Y. Zheng, et al., J. Am. Chem. Soc. 138 (2016) 7429-7435. DOI:10.1021/jacs.6b04031 |
| [46] |
Z.M. Wu, S.Z. Liu, X.Z. Cheng, et al., Chin. Chem. Lett. 27 (2016) 1731-1739. DOI:10.1016/j.cclet.2016.04.024 |
| [47] |
Y. Huang, W.H. Feng, Chin. Chem. Lett. 27 (2016) 357-360. DOI:10.1016/j.cclet.2015.11.012 |
| [48] |
C.L. Zhang, S. Liu, X.C. Liu, et al., Chin. Chem. Lett. 28 (2017) 1523-1527. DOI:10.1016/j.cclet.2017.03.010 |
| [49] |
C.C. Chen, S. Gao, Q. Qiu, Chin. Chem. Lett. 29 (2018) 1135-1138. DOI:10.1016/j.cclet.2018.01.005 |
| [50] |
B.L. Pentelute, Z.P. Gates, V. Tereshko, et al., J. Am. Chem. Soc. 130 (2008) 9695-9701. DOI:10.1021/ja8013538 |
| [51] |
K. Mandal, B.L. Pentelute, V. Tereshko, et al., J. Am. Chem. Soc. 131 (2009) 1146-1154. DOI:10.1002/pro.127 |
| [52] |
A.J. McCoy, R.W. Grosse-Kunstleve, P.D. Adams, et al., J. Appl. Crystallogr. 26 (1993) 658-674. DOI:10.1107/S0907444902016657 |
| [53] |
E.R. Zuiderweg, D.G. Nettesheim, K.W. Mollison, et al., Biochemistry 28 (1989) 172-185. DOI:10.1021/bi00427a025 |