 2020, Vol. 31
2020, Vol. 31 


b State Key Laboratory for Oxo Synthesis and Selective Oxidation, Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences, Lanzhou 730000, China
Organophosphine compounds are common in medicinal chemistry and biochemistry [1]. Among the members of the organophosphine family, hydroxyphosphoric acids display the unique biological activitives, and they have some attractive prospects as clinical drug molecules (Scheme 1a) [2]. Therefore, the development of effective and simple tactics to synthesize the hydroxyphosphoric acid or its precursor has attracted much attention [3]. So far, great efforts have been devoted to construct C—P bond including metal/photo-catalyzed C—P bond formation [3]. The common point of these two tactics is the way to form C—P bond through the phosphorus radical intermediate [3h, 3k]. While, the main drawbacks of these synthesis strategies were using the stoichiometric base or stoichiometric oxidant to complete the transformation [3j]. And more, the high temperature was essential to promote the reaction, especially for the metal-catalyzed reactions [3g].

|
Download:
|
| Scheme 1. Photoredox catalysis for the synthesis of γ-oxo-phosphonates. | |
In 2014, Ji group reported the method for the synthesis of γ-oxo-phosphine oxide by using stoichiometric Ag salt to introduce the phosphorus radical involved semipinacol rearrangement process, while the high reaction temperature was essential [4]. In the same year, Wu group fulfilled the similar transformation by using Ag salt as catalyst [5]. Last year, Zhang group successfully prepared various γ-oxo-phosphine oxides through photo-catalyzedreaction under mild condition, but external oxygen was essential to complete the catalytic cycle and the phosphorus precursors were limited to phosphine oxides [6]. Our group has always been focused on the chemistry of phosphorus, and hope to construct C—P bond under mild conditions without external oxidants either for metalcatalyzed reaction or photo-catalyzed reaction [3h, 3k, 3s, 3u]. In 2018, we discovered that the oxime phosphonates were easily converted to P radical under visible-light irradiation by using fac-Ir(ppy)3 as photo-catalyst, therefore constructing the new C—P bond [3k]. So, we questioned whether it would be possible to form γ-oxo-phosphonates, which could be easily converted to hydroxyphosphoric acid by using this oxime phosphonates as phosphorus radical precursor through photo-catalyzed reaction. Herein, we reported the method for the synthesis of γ-oxo-phosphonates via phosphorus radical involved semipinacol rearrangement process. Most important, this transformation is avoid of the external oxidants, and occurs very well under the sunlight irradiation, meanwhile the γ-oxo-phosphonate was easily derivatized toobtain γ-hydroxyphosphoric acid, thus highlights the synthesis value of this method.
In the initial study, we first selected 1, 1-diphenylprop-2-en-1-ol (1a) [8] and oxime phosphonate (2a) as substrates and fac-Ir(ppy)3 as photo catalyst to explore various solvents irradiated with 5 W blue LEDs. We were delighted to obtain the desired product 3a in 11% and 61% yield by using CH3CN and dioxane as the solvent (Table 1, entries 2 and 3). According to the previous work, we increased the reaction concentration to 0.2 mol/L, and the yield was up to 84% (Table 1, entries 4 and 5). Decreasing the loading of fac-Ir(ppy)3 or changing the proportion of the two substrates was not helpful (Table 1, entries 6 and 7). Finally, control experimental run in the absence of either photo catalyst or a visible light source did not give the desired product 3a (Table 1, entries 8 and 9).
|
|
Table 1 Investigation of the reaction conditions.a |
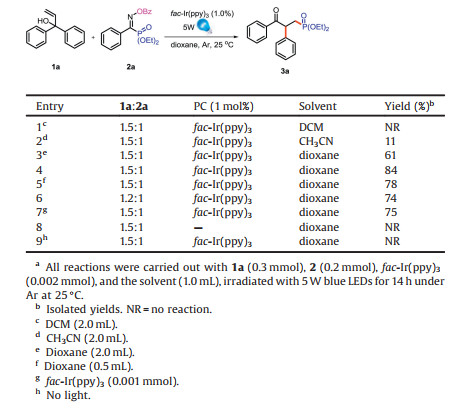
{kind=link}
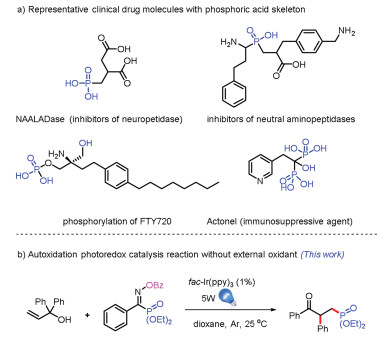
Having identified optimal conditions, a wide range of oxime phosphonates were submitted to this photo catalysis protocol (Scheme 2) [3k]. First, we investigated the influence of OPG group. When the OBz was changed to OAc or OPiv, we were delighted that the desired product could be obtained in 64% or 47% yields (2b and 2c). Next, we investigated the influence of substituent group on phosphorus atom (methoxy or isopropyl), we were also able to obtain the target product in moderate to excellent yields (2d, 61%; 2e, 81%). Most important, the R2 substituent was changed to alkyl group, the reaction also proceeded smoothly (2f-2i, 12%-58%). Furthermore, both electron-withdrawing groups (2l, 2m, 2p) and electron-donating group (2j, 2k, 2n, 2o) could be present on each aryl group (R2 and OBz), and good to excellent yields of 57% to 83% were obtained. These results demonstrated that the reaction tolerated various of oxime phosphonates very well.

|
Download:
|
| Scheme 2. Substrate scope for oxime phosphonates. All reactions were carried out with 1a (0.3 mmol), 2 (0.2 mmol), fac-Ir(ppy)3 (0.002 mmol), and dioxane (1.0 mL), irradiated with 5 W blue LEDs for 14 h under Ar at 25 ℃. Isolated yields. | |
{kind=link}
We then investigated the use of substituted allylic alcohol substrates. As shown in Scheme 3, when electron-donating groups (methyl or methoxy group) were introduced to the para-position of allylic alcohol, the yields dropped to 78% and 54% (3b and 3c). These results may be caused by the more electron-rich aryl group was more difficult to migrate compare to benzene group. To our delight, for the more electron-rich aryl group, we could also obtain the desired product in 48% yield (3g). Next, when electronwithdrawing groups (CF3, F, Cl) were introduced to the para-position of allylic alcohol, and moderate to good yields of 69% to 81% were obtained. Furthermore, when methyl group or methoxy group was introduced to the orth-position of allylic alcohol, good yields of 73% to 76% were obtained (3h and 3i). And more, for the meta-position of allylic alcohol, either electron-withdrawing group (3l) or electron-donating group (3j and 3k) could be tolerated, and good yields of 63% to 76% could be obtained. Moreover, good yields of 65% could be obtained for the nonterminal allylic alcohol substrate 3m.

|
Download:
|
| Scheme 3. Substrate scope for allylic alcohols. All reactions were carried out with 1a (0.3 mmol), 2 (0.2 mmol), fac-Ir(ppy)3 (0.002 mmol), and dioxane (1.0 mL), irradiated with 5 W blue LEDs for 14 h under Ar at 25 ℃. Isolated yields. | |
{kind=link}
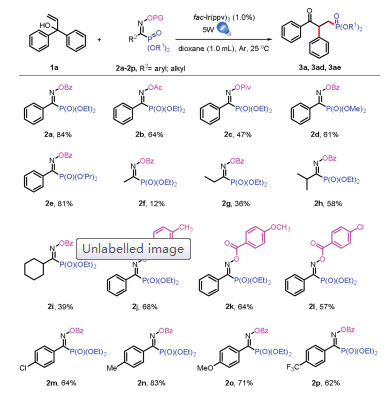
Next, to understand the influence of migration caused by the electronic nature of aryl group, unsymmetrical allylic alcohols carrying two different aryl groups were investigated (Scheme 4, 3n-3u). We were delighted that the reactions proceeded in good yields of 81% and 80% when thiophene and pyridine were used instead of benzene group (3n, 3o, 3o'). And more, we could get two isomers, which could be separated by chromatography for the pyridine case. These results demonstrated that the more electrondeficient aryl group migrated preferentially [6]. It was noteworthy that methyl group was introduced to the orth-position of one benzene group, the single isomer could be obtained in good yield of 71% (3p). More important, not only high regioselectivity but also functional-group tolerance was demonstrated for the successful realization of 3u with a lower yield of 29%.

|
Download:
|
| Scheme 4. Substrate scope for unsymmetrical allylic alcohols.All reactions were carried out with 1a (0.3 mmol), 2 (0.2 mmol), fac-Ir(ppy)3 (0.002 mmol), and dioxane (1.0 mL), irradiated with 5 W blue LEDs for 14 h under Ar at 25 ℃. Isolated yields. | |
{kind=link}
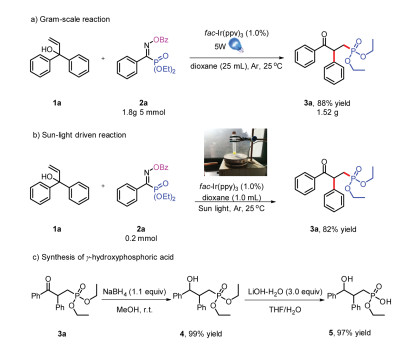
Several experiments were implemented to highlight the synthesis value of this method. The reaction proceeded very well with gram-scale yield of 88% (Scheme 5a). Furthermore, when the sunlight was used instead of 5 W blue LEDs, the desired product could also be obtained with 82% yield (Scheme 5b). Meanwhile the γ-oxo-phosphonate was easily derivatized to obtain γ-hydroxyphosphoric acid 5 (Scheme 5c) [9]. These results highlight the synthesis value of this tactic in drug discovers.

|
Download:
|
| Scheme 5. Applied experiments. | |
{kind=link}
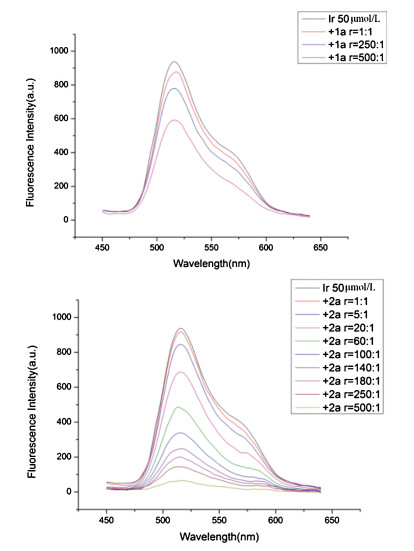
In the presence of 2, 2, 6, 6-tetramethylpiperidine-1-oxyl (TEMPO, 2.0 equiv.) or 2, 6-di-tert-butyl-4-methylphenol (BHT, 2.0 equiv.), the observed inhibition of thereaction indicated that a radical process might be involved (Scheme S1 in Supporting information) [10]. According to our previous work, oxime phosphonate could be a suitable oxidant to be reduced by *Ir (Ⅲ), therefore converting to the active P radical [3k]. So, fluorescence spectra were collected on Cary Eclipse Fluorescence Spectrophotometer. A significant decrease of fac-Ir(ppy)3 luminescence was successfully observed in the presence of 2a (Fig. 1). This result suggested that it was the substrate 2a that quenched the excited photocatalyst *Ir(Ⅲ) [7].

|
Download:
|
| Fig. 1. Luminescence quenching experiments of 1a (top) and 2a (bottom). | |
{kind=link}
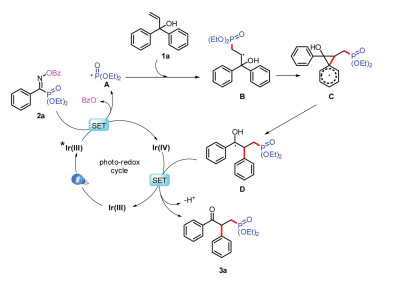
Based on the experimental results of the mechanistic investigation and previous mechanistic studies, details of the mechanism of the proposed phosphorus radical involved semipinacol rearrangement reaction are outlined in Scheme 6. Irradiation of photo catalyst fac-Ir(ppy)3 Ir(Ⅲ) with visible light leads to the formation of excited-state *Ir(Ⅲ), which could be oxidized by oxime phosphonate 2a with producing the P radical A [3k]. Then P radical A is rapidly trapped by alkene of the substrate 1a to generate alkyl radical B, which immediately forms benzyl radical D through semipinacol rearrangement process. Finally, we expected that oxidation of the resulting benzyl radical D should generate benzyl cation with deprotonation to give the final product 3a and regenerate the ground-state photo catalyst Ir(Ⅲ), completing the proposed catalytic cycle.

|
Download:
|
| Scheme 6. Proposed mechanistic pathway. | |
{kind=link}
In summary, we have demonstrated a new, mild approach for the synthesis of γ-oxo-phosphonates (the precursor of hydroxyphosphoric acid) through the semipinacol rearrangement tactics involved the photo-induced phosphorus radical process. Most important, this transformation is avoid of the external oxidants, and occurs very well under the sunlight irradiation, meanwhile the γ-oxo-phosphonate was easily derivatized to obtain γ-hydroxyphosphoric acid, thus highlights the synthesis value of this method.
AcknowledgmentsWe are grateful for the National Natural Science Foundation of China (No. 21532001) and International Joint Research Centre for Green Catalysis and Synthesis, Gansu provincial Sci. & Tech. Department (No. 2016B01017) for financial support.
Appendix A. Supplementary dataSupplementary material related to this article can befound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.08.011.
| [1] |
(a) P.J. Murphy, Organophosphorus Reagents, OxfordUniv. Press, Oxford, 2004; (b) D.T. Kolio, Chemistry and Application of H-phosphonates, Elsevier Science, Oxford, 2006; (c) N.S. Li, J.K. Frederiksen, J.A. Piccirilli, Acc. Chem. Res. 44 (2011) 1257-1269; (d) H.H. Chen, L. Zhu, K.B. Zhong, et al., Chin. Chem. Lett. 29 (2018) 1237-1241. |
| [2] |
(a) P.F. Jackson, K.L. Tays, K.M. Maclin, et al., J. Med. Chem. 44 (2001) 4170-4175; (b) R. Albert, K. Hinterding, V. Brinkmann, et al., J. Med. Chem. 48 (2005) 5373-5377; (c) S. Vassiliou, E.W. Tomczak, L. Berlicki, et al., J. Med. Chem. 57 (2014) 8140-8151; (d) Z. Novakova, K. Wozniak, A. Jancarik, et al., J. Med. Chem. 59 (2016) 4539-3550; (e) G.P. Horsman, D.L. Zechel, Chem. Rev. 117 (2017) 5704-5783. |
| [3] |
(a) A.L. Schwan, Chem. Soc. Rev. 33 (2004) 218-224; (b) C.S. Demmer, N.K. Larsen, L. Bunch, Chem. Rev. 111 (2011) 7981-8006; (c) X.Q. Pan, J.P. Zou, W.B. Yi, W. Zhang, Tetrahedron 71 (2015) 7481-7529; (d) K. Luo, W.C. Yang, L. Wu, Asian J. Org. Chem. 6 (2017) 350-367; (e) B.G. Cai, J. Xuan, W.J. Xiao, Sci. Bull. 64 (2019) 337-350; (f) Y.L. Zhang, K. Sun, Q.Y. Lv, et al., Chin. Chem. Lett. 30 (2019) 1361-1368; (g) C.G. Feng, M. Ye, K.J. Xiao, S. Li, J.Q. Yu, J. Am. Chem. Soc. 135 (2013) 9322-9325; (h) Y.M. Li, M. Sun, H.L. Wang, Q.P. Tian, S.D. Yang, Angew. Chem. Int. Ed. 52 (2013) 3972-3976; (i) L.L. Liao, Y.Y. Gui, X.B. Zhang, D.G. Yu, Org. Lett. 19 (2017) 3735-3738; (j) Y.H. Li, Y.Y. Zhu, S.D. Yang, Org. Chem. Front. 5 (2018) 822-826; (k) C. Li, Z.C. Qi, Q. Yang, X.Y. Qiang, S.D. Yang, Chin. J. Chem. 36 (2018) 1052-1058; (l) R. Isshik, K. Muto, J. Yamaguchi, Org. Lett. 20 (2018) 1150-1153; (m) J. Yang, T.Q. Chen, L.B. Han, J. Am. Chem. Soc. 137 (2015) 1782-1785; (n) D.P. Hari, B. König, Org. Lett. 13 (2011) 3852-3855; (o) Y.L. Zhao, G.J. Wu, F.S. Han, Chem. Commun. 48 (2012) 5868-5870; (p) V. Qunit, F. Morlet-Savary, J.F. Lohier, et al., J. Am. Chem. Soc. 138 (2016) 7436-7441; (q) L.B. Niu, J.M. Liu, H. Yi, et al., ACS Catal. 7 (2017) 7412-7416; (r) L.B. Niu, H. Yi, S.C. Wang, et al., Chem. Commun. 54 (2018) 1659-1662; (s) C.H. Wang, Y.H. Li, S.D. Yang, Org. Lett. 20 (2018) 2382-2385; (t) Y.Y. Song, L.L. Wang, Z. Duan, F. Mathey, Chin. Chem. Lett. 31 (2020) 329-332; (u) Y.N. Ma, S.X. Li, S.D. Yang, Acc. Chem. Res. 50 (2017) 1480-1492. |
| [4] |
X.Q. Chu, Y. Zi, H. Meng, X.P. Xu, S.J. Ji, Chem. Commun. 50 (2014) 7642-7645. DOI:10.1039/c4cc02114b |
| [5] |
X. Mi, C. Wang, M. Huang, Y. Wu, Y.J. Wu, Org. Biomol. Chem. 12 (2014) 8394-8397. DOI:10.1039/C4OB01739K |
| [6] |
Y. Yin, W.Z. Weng, J.G. Sun, B. Zhang, Org. Biomol. Chem. 16 (2018) 2356-2361. DOI:10.1039/C8OB00231B |
| [7] |
X.Y. Yu, J.R. Chen, W.J. Xiao, et al., Angew. Chem. Int. Ed. 57 (2018) 738-743. DOI:10.1002/anie.201710618 |
| [8] |
(a) J.M. Fan, C.F. Wan, Wang Q, et al., Org. Biomol. Chem. 7 (2009) 3168-3172; (b) F.F. Mo, L.J. Trzepkowski, G.B. Dong, Angew. Chem. Int. Ed. 51 (2012) 13075-13079; (c) Z. Zhang, C. Li, S.H. Wang, et al., Org. Biomol. Chem. 15 (2017) 3239-3247. |
| [9] |
(a) J. Desroches, P.A. Champangne, Y. Behassine, J.K. Paguin, Org. Biomol. Chem. 13 (2015) 2243-2246; (b) T.W. Bevan, J.F. Taylor, H. Wong, D.T. Northcote, J.E. Harvey, Tetrahedron 74 (2018) 2942-2955. |
| [10] |
(a) E.B. Jane, R.S. Davidson, Makromol. Chem. Rapid. Commun. 8 (1987) 311-314; (b) A.P. William, T.G. Jen, D.F. Church, J. Org. Chem. 50 (1985) 185-189. |