 2020, Vol. 31
2020, Vol. 31 


Helical chirality is unique for the non-existence of stereogenic centers and has drawn great interests of scientists to build artificial chiral helix since Pauling first proposed α-helix [1], and Watson and Crick discovered the double helical structure for DNA [2] in the early 1950s. To mimic the helical structure of DNA, a number of artificial folded molecules termed as foldamers have been reported in the past few decades [3]. Aromatic foldamers bearing helical conformation driven by non-covalent forces are one of intensively studied foldamers, which exhibit an equilibrium between the left (M)- and right (P)- handed enantiomers. Breaking of such equilibrium by attaching chiral moieties to aromatic foldamers generates chiral foldamers with a preferred one-handedness [4], leading to many potential applications in chiral recognition [5], chiral electrooptical devices and asymmetric catalysis. Although the inductions of helicity in foldamers can be readily achieved by appending chiral motifs at the end of foldamers, inserting chiral unit into the middle of folded sequences, or binding chiral guests in helical cavities, the elegant examples on quantitative chiral inductions have been reported in only a few cases [6]. To some extent, lack of absolute control of helical handedness in chiral foldamers would greatly limit their applications in dissymmetric catalysis [7], photocatalysts [8], storage devices [9], and circularly polarized luminescence (CPL) laser [10].
We have previously achieved the absolute control of helical sense in quinoline oligoamide foldamers (QOFs) by appending β-pinene-derived pyridyl moiety at the N-terminus [6c], or locking the dynamic interconversion between the M- and P-conformers [11]. Very recently, we have reported the quantitative induction of one-handed helicity in QOFs by introducing chiral oxazolylanilines at the C-terminus [12]. These QOFs with absolute one-handedness exhibited strong CPL signals and high luminescence dissymmetric factors glum, which display the significant dependence on the length of QOFs. To further understand the quantitative induction of one-handed helicity caused by oxazolyanilines, we designed and synthesized tetrameric and octameric QOFs containing different residues in oxazolylanilines and explored the effect of the sizes of the residues on the chiral induction in these QOFs.
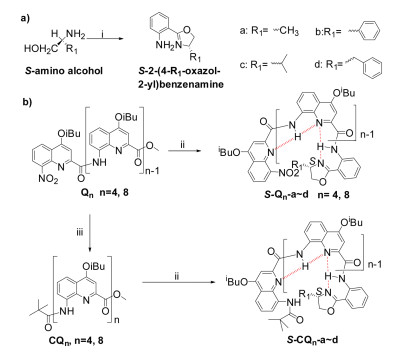
Previously, Ivan and co-workers found that the steric effect of chiral group at the C-terminus of QOFs plays a dominant role in amplifying the chirality in the case of incomplete chiral inductions, where the bulkiest group in the chiral center tends to point away from the helix while the smallest one prefers to point to the helix [4b]. To explore if the steric effect in the chiral centers would also influence the chiral induction of QOFs in the case of absolute chiral inductions, chiral oxazolylanilines with the different residues ranging from methyl to benzyl substitutes (Fig. 1) were designed. We expected there is a very stable intramolecular hydrogen bond network between QOFs and chiral induced group that would efficiently prevent the rotation of chiral induced groups and significantly eliminate the effect of the residues in the chiral induced groups on the chiral inductions. The synthetic routes of the tetrameric or octameric QOFs containing various chiral oxazolylaniline motifs were outlined in Scheme 1. First of all, chiral oxazolylanilines were prepared by treating 2-aminobenzonitrile and enantiomeric 2-aminoalcohols in anhydrous chlorobenzene with zinc chloride as catalysts under refluxing condition according to the reported methods [13]. The compounds S-Qn-a~d were obtained by coupling corresponding quinoline tetrameric or octameric acids with chiral oxazolylanilines with the yield of over 80% for tetrameric QOFs and over 60% for octameric QOFs, respectively.

|
Download:
|
| Fig. 1. Parts of the 1H NMR spectra of (a) S-Q4-a, (b) S-Q4-b, (c) S-Q4-c, (d) S-Q4-d, (e) S-Q8-a, (f) S-Q8-b, (g) S-Q8-c and (h) S-Q8-d in CDCl3 at 298 K. | |
The 1H NMR spectra of tetramers S-Q4-a~d feature a single set of signals in CDCl3 at 298 K and the sharp signals appearing at 11.5~13.5 ppm are assignable to the carboxamide protons. Thereinto, the chemical shifts appearing at 13.0~13.3 ppm can be assigned to the amide protons close to the oxazolylanilino groups according to our previous results [12], which are about 1 ppm lower than those located inside the helix, owing to the stable three-center hydrogen bonding networks and the weaker effects of π-π aromatic stackings compared with those formed between quinoline rings. Furthermore, the signals of the amide protons close to the oxazolylamine groups containing alkyl substituents in S-Q4-a and S-Q4-c appear at 13.13 and 13.18 ppm, respectively, which are about 0.1 ppm lower than those containing aromatic substituents in S-Q4-b and S-Q4-d, whose protons appear at 13.03 ppm. The nuanced differences in the chemical shifts are presumablyattributed to the shielding effect of aromatic rings in the chiral centers. Additionally, considering that there maybe the possibility that the proton signals coalesce due to a fast interconversion between S—P and S—M diastereoisomeric QOF tetramers at 298 K on the NMR time scale [14], variable low temperature 1H NMR experiments were carried out. As we expected, the spectra of all tetramers exhibited single set of peaks and no split signals was observed even at 183 K (Figs. S5 and S6 in Supporting inforamtion). The observations support the facts that chiral inductions of S-Q4-a~d are quantitative and the diastereomeric excess (de) in each case is ≥ 99%. As for S-Q8-a~d, the chemical shifts exhibit similar features to those of S-Q4-a~d, but appear in the higher field because of stronger shielding effect of aromatic rings in octamers. We believe that the absolute controlling of helicity can be accounted for by the stable hydrogen bonding network forming between the oxazolylanino group and quinoline amide.
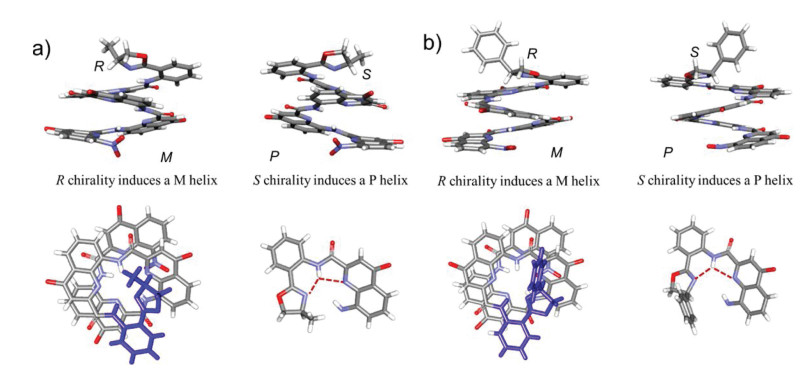
To further conform the presence of the three-center hydrogen bonding network mentioned above, the racemates (±)S/R-Q4-a and (±)S/R-Q4-b were designed and synthesized. Their crystals were grown by diffusing hexane into a chloroform concentrated solution. Both crystal structures of (±)S/R-Q4-a and (±)S/R-Q4-b display the formation of a three-center hydrogen bonding network between the amide hydrogen and N atoms in both oxazolylaniline and adjacent quinoline ring (Fig. 2), which renders oxazolylanilino group to adopt a parallel conformation to the adjacent quinoline rings in the helix. This parallel conformation leads to that the methyl or phenyl group in chiral center is almost perpendicular to the helix and points away from the helix to minimize the steric barrier, while the proton points to the helix. The space groups of (±)S/R-Q4-a and (±)S/R-Q4-b are P-1 and P21/n, respectively. More importantly, XRD data also reveal that one pair of enantiomers cocrystallize as true racemates in which the S-chiral center always induces the P helicity, while the R-chiral center always induces the M helicity (Fig. 2), consistent with our previous reports [4b].

|
Download:
|
| Fig. 2. The crystal structures of (a) the side and top views of racemic (±)S/R-Q4-a and its three-center hydrogen bonding network and (b) the side and top views of racemic (±) S/R-Q4-b and its three-center hydrogen bonding network. In the both cases, R chiral center always induces a M helix and S chiral center always induced a P helix. The oxazolylanilino groups were labeled in blue in the top views. The isobutyl groups were deleted for clarity. | |
As shown in Fig. 3, the CD spectra of S-Q4-a~d featured almost identical and exhibited positive intense cotton effect at 250–450 nm region originated from quinoline chromophores, indicating a preferred P-handedness. S-Q4-a~d displayed peaks at 386 nm with Δε around 93 L·mol-1 cm-1. The absorption dissymmetric factors gabs, defined as 2(εL-εR)/(εL+εR) [15], were calculated to be around 0.020 (Table S1 in Supporting information). The CD spectra of S-Q8-a~d also exhibited positive cotton effects at 250~450 nm region similar to that of S-Q4-a~d, but with larger CD intensity. The gabs values of S-Q8-a~d were found in the range from 0.028 to 0.034 (Table S1), which are apparently larger than those of S-Q4-a~d. The enhanced gabs values are beneficial from the increase on the length of the foldamers. The above results clearly demonstrate that the central chirality of the oxazolylanilino chiral motifs has been efficiently transferred into the helicity of the QOFs without depreciation of chiral information, leading to the quantitative chiral inductions. The absolute controlling of helicity is attributed to the stable three-center hydrogen bonding network between the chiral group and foldamer. We also note that the substituents in the chiral centers have negligible effect on chiral inductions, which is completely different from the results observed in the case of incomplete chiral induction [4b].

|
Download:
|
| Fig. 3. (a). CD spectra of S-Q4-a~d and S-Q8-a~d (1.0 × 10-5 mol/L, DCM, 298 K); (b). UV–vis spectra of S-Q4-a~d and S-CQ8-a~d (1.0 × 10-5 mol/L, DCM, 298 K). | |
On the other hand, materials with the feature of circularly polarized emission have attracted growing attention due to its potential applications in chiroptical or electrooptical devices and information storage [8b, 8c, 9a]. In order to shed light on chiral conformational and three-dimensional information of the luminescent QOFs in the excited state, CPL active S-CQn-a~d were prepared by simple coupling the pivaloyl protected QOFs acids with chiral oxazolylanilines with an overall yield of 64%~86% (Scheme 1). 1H NMR studies reveal that the spectra of S-CQ4-a~d and S-CQ8-a~d feature as single set of signals as its nonluminescent counterparts S-Qn-a~d do, indicative of quantitative chiral induction for S-CQn-a~d.

|
Download:
|
| Scheme 1. Synthetic route for S-Qn-a~d and S-CQn-a~d. Reaction conditions: ⅰ. 2-aminobenzonitrile (1.0 equiv.), S-amino alcohols (1.5 equiv.), anhydrous ZnCl2 powder (2.5 equiv.), chlorobenzene, reflux, 36 h; ⅱ. (a) NaOH (2.5 equiv.), THF/MeOH/H2O (v/v/v = 10:2:1), 40 ℃; (b) Oxalyl chloride (2.0 equiv.), DCM, room temperature, 2 h; (c) S-chiral amines (0.9 equiv.), DIPEA (2.0 equiv.), DCM; ⅲ. (a) Pd/C (0.1 equiv.), HCOONH4, NH4VO3, EA/EtOH/H2O (v/v/v = 4:1:0.5); 95 ℃; (b) Pivaloyl chloride (1.2 equiv.), DIEPA (2.0 equiv.), DCM, room temperature. | |
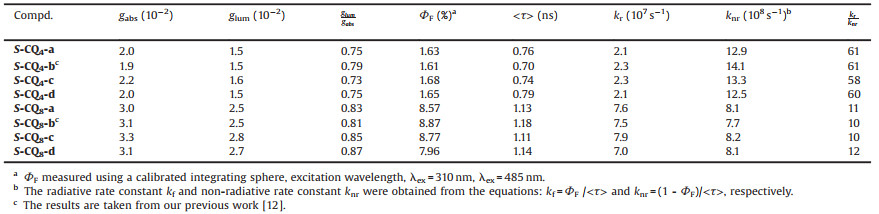
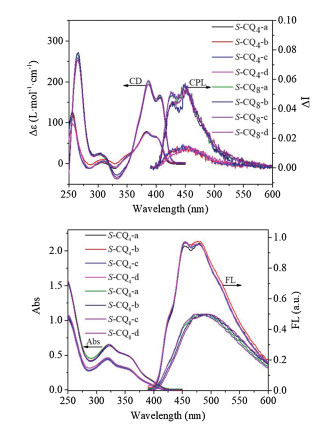
Before we set off to evaluate chiral properties of S-CQn-a~d in the excited state, the properties in the ground state of S-CQn-a~d were investigated by CD. As shown in Figs. 3 and 4, it is not surprised to find that S-CQ4-a~d display the P helicity with the absorption dissymmetric factors gabs almost identical to its non-luminescent counterparts S-Q4-a~d (Table 1 and Table S1), hinting that the modifications at N-terminal exert no effect on the absorption dissymmetric factors inthe ground state. The emission of S-CQ4-a~d landed at the blue region, exhibiting its maximal emission at around 480 nm. The fluorescence quantum yields were determined to be about 1.61% using a calibrated integrating sphere (Table 1). The fluorescent properties of S-CQ4-a~d allow us to investigate the chiral induction at the excited state. As shown in Fig. 4, the CPL spectra display the bands around 400~600 nm, in accordance with that of the fluorescent spectra. Moreover, all S-configured QOFs exhibit positive CPL signals, indicative of aP helical sense in each case that is consistent with the observations in the CD experiments. The emission dissymmetry factors glum were calculated according to the definition of glum = 2(IL-IR)/(IL+IR) [15a], where IL and IR refer to left- and right-handed circularly polarized light, respectively [15a]. The glum values of S-CQ4-a~d are about 0.02, which are almost same (Table 1), implying that the substituents in the chiral centers exert no effect on the glum values.

|
Download:
|
| Fig. 4. (a) CD and CPL spectra of S-CQ4-a~d and S-CQ8-a~d; (b) UV–vis and FL spectra of S-CQ4-a~d and S-CQ8-a~d (1.0 × 10-5 mol/L, DCM, 298 K). | |
|
|
Table 1 The summery of the chiroptical characters of compounds S-CQn-a~d. |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The steady-state fluorescence spectra of S-CQ8-a~d show blue shift emission with a maximal peak at 455 nm and the fluorescence quantum yields were determined to be about 8% (Table 1). The CD and CPL studies reveal similar chiroptical properties to those of its short analogs S-CQ4-a~d. However, their gabs and glum values were determined to be about 0.031 and 0.027, respectively, which are apparently larger than the corresponding values for the short S-CQ4-a~d (Table 1) and those for helically chiral molecules reported to date [16]. The large glum values are probably attributed to the increase on the lengths of QOFs. Besides, the ratios of emission and absorption dissymmetric factors glum/gabs range from 0.73 to 0.79 for S-CQ4-a~d and from 0.81 to 0.87 for S-CQ8-a~d, respectively, suggesting that the dissymmetry of these chiral QOFs is mainly kept in the excited state [16c].
Fluorescence decay measurements were conducted using a nanosecond pulsed laser system in DCM at room temperature. The decays of S-CQ4-a~d follow first-order kinetics with lifetimes (τ) being 0.70 ~ 0.79 ns at 485 nm when excited at 360 nm (Table 1 and Table S2). However, the longer QOFs S-CQ8-a~d display the similar single-exponential decays with longer τ = 1.11–1.18 ns at 455 nm when excited at 360 nm. The longer fluorescence lifetimes of S-CQ8-a~d compared to S-CQ4-a~d are also accounted for by the contribution of the longer lengths of QOFs. Additionally, the ratios of kr/knr of S-CQ8-a~d are 10–12, much smaller than those of S-CQ4-a~d (58–61), stating that with the lengths of chiral QOFs increases, the excitedstate prefer to decay via radiative channel, probably due to the more rigid structure in the excited state of octamers.
In summary, we synthesized a series chiral QOFs containing chiral oxazolylanilino motifs that absolutely control the helicities of QOFs by the stable three-center hydrogen-bonding network between the chiral motif and QOF, which restricts the rotation of the chiral group and eventually lead to complete chiral induction. Both the absorption and emission dissymmetric factors of S-CQn-a~d are almost identical respect to the corresponding length of S-CQn-a~d, regardless of the sizes of the substituents in the chiral oxazolylaniline. However, the chemical shift exhibit nuance difference, due to the different electronic featuresof the substituents in the chiral centers. Furthermore, the dissymmetry factors increase with the elongations of the lengths of QOFs. The absolute control of helicity presented here provides a new method for improving the dissymmetry factors for designing advanced organic molecules bearing a helical conformation with unprecedent chiroptical properties.
AcknowledgmentsWe acknowledge the financial supports from the 973 Program (No. 2015CB856502) and the National Natural Science Foundation of China (No. 21672026). We also thank Applied Photophysics for their generous assistance on CPL measurements.
Appendix A. Supplementary dataSupplementary material related to this article can befound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.07.061.
| [1] |
L. Pauling, R.B. Corbey, H.R. Branson, Natl. Acad. Sci. U. S. A. 378 (1951) 205-211. |
| [2] |
J.D. Watson, F.H.C. Crick, Nature 171 (1953) 737-738. DOI:10.1038/171737a0 |
| [3] |
(a) Z.T. Li, J.L. Hou, C. Li, H.P. Yi, Chem. -Asian J. 1 (2006) 766-778; (b) D.W. Zhang, X. Zhao, J.L. Hou, Z.T. Li, Chem. Rev. 112 (2012) 5271-5316. |
| [4] |
(a) H. Jiang, C. Dolain, J.M. Leger, et al., J. Am. Chem. Soc. 126 (2004) 1034-1035; (b) C. Dolain, H. Jiang, J.M. Leger, I. Huc, J. Am. Chem. Soc. 127 (2005) 12943-12951; (c) F.S. Bie, Y. Wang, J. Shang, et al., Eur. J. Org. Chem. 8 (2013) 8135-8144; (d) H. Jiang, Q.L. Li, G.X. Wang, Chin. J. Org. Chem. 38 (2018) 1065-1084. |
| [5] |
C. Li, G.T. Wang, H.P. Yi, et al., Org. Lett. 9 (2007) 1797-1800. DOI:10.1021/ol070492l |
| [6] |
(a) A.M. Kendhale, L. Poniman, Z.Y. Dong, et al., J. Org. Chem. 76 (2011) 195-200; (b) Y. Ferrand, A.M. Kendhale, B. Kauffmann, et al., J. Am. Chem. Soc. 132 (2010) 7858-7859; (c) L. Zheng, Y.L. Zhan, C.Y. Yu, et al., Org. Lett. 19 (2017) 1482-1485; (d) H.Y. Hu, J.F. Xiang, Y. Yang, C.F. Chen, Org. Lett. 10 (2008) 1275-1278. |
| [7] |
I. Huc, H. Jiang, Organic foldamers and helices, in: J.W. Steed, P.A. Gale (Eds.), Supramolecular Chemistry: From Molecules to Nanomaterials, John Wiley & Sons Ltd, Chichester, 2012, pp. 2183-2206.
|
| [8] |
(a) N. Tamaoki, M. Wada, J. Am. Chem. Soc. 128 (2006) 6284-6285; (b) L.B. Wang, L. Yin, W. Zhang, et al., J. Am. Chem. Soc. 139 (2017) 13218-13226; (c) G. Yang, L. Zhu, J. Hu, et al., Chem. -Eur. J. 23 (2017) 8032-8038; (d) Y. Lu, L.L. Zhu, Chin. Chem. Lett. 29 (2018) 1591-1600. |
| [9] |
(a) C.S. Wang, H.S. Fei, Y. Qiu, et al., Appl. Phys. Lett. 74 (1999) 18-21; (b) A. Natansohn, P. Rochon, Chem. Rev. 102 (2002) 4139-4175; (c) D. Sankar, P.K. Palanisamy, S. Manickasundaram, P. Kannan, Opt. Mater. (Amst) 28 (2005) 1101-1107. |
| [10] |
J. Jimenez, L. Cerdan, F. Moreno, et al., J. Phys. Chem. C 121 (2017) 5287-5292. DOI:10.1021/acs.jpcc.7b00654 |
| [11] |
L. Zheng, C.Y. Yu, Y.L. Zhan, et al., Chem. -Eur. J. 23 (2017) 5361-5367. DOI:10.1002/chem.201700134 |
| [12] |
D. Zheng, L. Zheng, C.Y. Yu, et al., Org. Lett. 21 (2019) 2555-2559. DOI:10.1021/acs.orglett.9b00450 |
| [13] |
M. Luo, Curr. Org. Synth. 12 (2015) 660-672. DOI:10.2174/157017941205150821153905 |
| [14] |
H. Jiang, J.M. Leger, C. Dolain, et al., Tetrahedron 59 (2003) 8365-8374. DOI:10.1016/j.tet.2003.08.058 |
| [15] |
(a) J.P. Riehl, F.S. Richardson, Chem. Rev. 86 (1986) 1-16; (b) H. Anetai, T. Takeda, N. Hoshino, et al., J. Phys. Chem. C 122 (2018) 6323-6331. |
| [16] |
(a) E.M. Sanchez-Carnerero, A.R. Agarrabeitia, F. Moreno, et al., Chem. -Eur. J. 21 (2015) 13488-13500; (b) H. Maeda, Y. Bando, K. Shimomura, et al., J. Am. Chem. Soc. 133 (2011) 9266-9269; (c) C.M. Cruz, S. Castro Fernandez, E. Macoas, et al., Angew. Chem. Int. Ed. 57 (2018) 14782-14786. |