 2020, Vol. 31
2020, Vol. 31 


Higher alcohols are considered as important compounds in chemical, biological and clean energy industries. Ethanol and isobutanol are excellent gasoline additives to raise the octane number and the combustion efficiency, and long-chain alcohols (C6-C22) can be used as intermediates to produce surfactants and detergents [1, 2]. All of these facts highlight the promising potential market for higher alcohol synthesis (HAS). With the gradual depletion of crude oil reserves and the increasing environmental issues, the utilization of syngas, which could be derived from coal, biomass and nature gas, has attracted considerable interest. The direct conversion of syngas to higher alcohols is recognized as a promising route with respect to its environmental friendliness and atom economy [3].
Co-based catalysts have been extensively investigated and used in Fischer-Tropsch synthesis (FTS) [4]. A lot of studies in HAS focused on the Cu modified Co-based bimetallic catalysts owing to their good catalytic performance [5, 6]. For Cu-Co based catalysts, Co is generally accepted to enable the CO dissociative adsorption and offer a carbon chain growth site, while Cu favors CO adsorption and insertion. The close contact between Cu and Co species is thus essential for HAS [7]. However, the bulk Cu-Co is unlikely to exist stably because of the low solubility between these two metals. And the phase separation between Cu and Co elements would result in the formation of methanol and hydrocarbons [8, 9]. Recently, it is proposed that Co0-Con+ presents an effective synergy as well, in which Con+ species act as the CO insertion site facilitating the molecular adsorption of CO [10, 11]. Chen et al. [12] obtained Co/CoOx interface by using the strong metal-support interaction between CeO2 support and Co sites. It is demonstrated that the formation of Co-CoOx pairs was critical to enhance the alcohol selectivity in HAS.
Significant researches have reported that the physical and chemical properties of the supports, such as morphology, porosity, and surface functional groups have a great influence on the metal sites and catalytic performance [13, 14]. Among all the carriers, carbon materials have attracted considerable attention due to their tunable porosity and characteristic surface chemistry [15, 16]. A lot of studies explored the effect of different types of carbon materials on active species. Zhang and coworkers [17] compared h-type carbon nanotubes (CNTs) and p-type CNTs supported Cu-Co based catalysts in HAS. The sample with h-type CNTs showed better catalytic activity because of its superior capability of activating H2, which increased the reaction rate of surface hydrogenation in HAS. Pei et al. [18] deposited Co sites on activated carbon materials derived from coconut shell (AC1) and almond nut (AC2). They found that significant amount of Co2C species existed in Co/AC1 catalyst, but there is no presence of Co2C observed in Co/AC2 catalyst. Hence, the modification of carbon supports is a possible way to provide suitable chemical environments for active sites in HAS, which may lead to further understandings between surface properties and catalytic behavior.
In this work, we synthesized a series of cobalt-based catalysts supported on carbon layer-coated ordered mesoporous silica (OMS) composites, in which glucose, furfuryl alcohol (FA) and 2, 3-dihydroxynaphthalene (DN) were used as carbon sources respectively. Based on the detailed characterization of composite support, we investigated the effect of surface properties on the dispersion of nanoparticles, Co0-Con+ species distribution and the corresponding catalytic behavior in HAS, which may provide new thoughts in rational design of catalysts.
OMS was synthesized by hydrothermal treatment according to the procedure reported by Zhao and coworkers [19].10 wt% carbon layer-coated OMS supports were prepared by the impregnation of precursors and subsequent carbonization. The detailed information of experiments is given in the Supporting information.
The thermal stability and carbon coating amount of x-10C/OMS supports were analyzed by TG, and the results are given in Fig. S1 (Supporting information). There is no obvious weight loss in the OMS support, indicating the completely decomposition of P123 after calcination. A rapid weight loss in the range of 450-700 ℃ can be observed in x-10C/OMS samples, which corresponds to the decomposition of carbon in air [20]. Notably, the carbon contents of Glucose-10C/OMS, FA-10C/OMS and DN-10C/OMS is 9.6 wt%, 9.7 wt% and 9.7 wt% respectively, which were calculated from the weight loss of TG curves. The results indicate that the carbon coating contents of all x-10C/OMS samples are similar and in good agreement with the theoretical value.
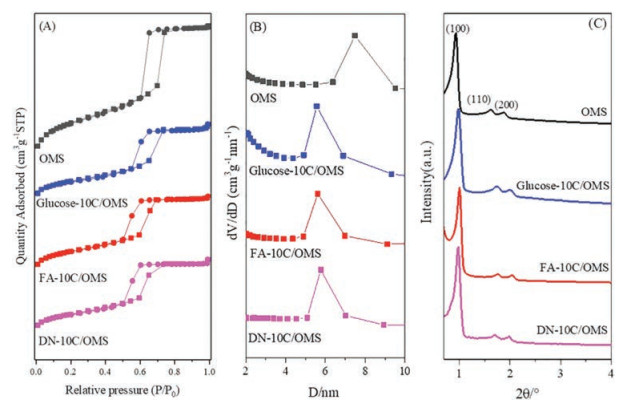
N2 adsorption-desorption isotherms of the x-10C/OMS supports are shown in Fig. 1A. All samples exhibit Langmuir type Ⅳ isotherms with an H1 hysteresis loop, corresponding to the typical mesoporous structure with uniform pore diameter [21]. The pore size distribution of x-10C/OMS are given in Fig. 1B and Table S1 (Supporting information). All carbon layer-coated samples exhibit similar pore size (about 5.7 nm), which is smaller than that of OMS (8.2 nm). It is demonstrated that the carbon layer can be uniformly coated inside the pores of OMS [22]. Moreover, the similar pore size of x-10C/OMS supports indicate that the coating thickness is basically consistent, which is independent of the different carbon source. In order to investigate the effect of carbon coating on the structure of OMS support, low-angle XRD patterns of x-10C/OMS supports were measured as well. As shown in Fig. 1C, the (100), (110) and (200) reflection peaks of OMS are clearly observed in all patterns. It indicates that the well-ordered p6mm symmetry can be maintained in all carbon layer-coated samples [19], which is consistent with the N2 adsorption-desorption results. In addition, it is noteworthy that all the three reflection peaks of OMS shifted to a higher angle after carbon layer coating. This shift is due to the thermal shrinkage during the heat-treatment step [23, 24].

|
Download:
|
| Fig. 1. N2 adsorption-desorption isotherms (A), pore size distribution (B) and low angle XRD patterns (C) of x-10C/OMS and OMS supports. | |
FT-IR spectroscopy was performed to measure the variation of the functional groups after carbon coating (Fig. S2 in Supporting information). The OMS sample show characteristic bands at 810 cm-1 and 1080 cm-1 corresponding to the asymmetric and symmetric stretching vibrations of Si-O-Si, as well as the bands at 950 cm-1 and 2600-3800 cm-1 caused by the stretching of Si-O-H [25]. After the coating of carbon layer, the characteristic bands assigned to the Si-O-H groups disappeared. Moreover, a new peak at 1610 cm-1 appeared in x-10C/OMS samples, which corresponds to C = C stretching vibrations [26]. This demonstrates that the surface of OMS are entirely coated by carbon layer. Nevertheless, owing to the overlap of the stretching vibrations between Si-O-Si and C-O band (1000-1500 cm-1) [27], the detailed information about the carbon surface were unavailable.
The carbon layer derived from different sources may present different surface properties. Here Raman spectra, zeta-potential and XPS were measured to investigate the effect of carbon source on the surface properties. The graphitization degree has been considered as an important feature of carbon materials, which provides valuable information about surface defects and graphitic characters [28]. Raman spectroscopy was used to characterize the graphitization degree of samples. As shown in Fig. S3 (Supporting information) and Table 1, the Raman spectra of x-10C/OMS supports exhibit two distinct peaks at 1337 cm-1 and 1585 cm-1. The peak at 1337 cm-1 corresponds to the D band, associated with the surface defects and disordered graphite species. The other peak at 1585 cm-1 is assigned to G band, resulting from the stretching vibration of C = C bond in typical graphitic layers. Typically, the ratio of the intensity of D band to G band reflects the defects and disordered degree of carbon materials [29]. We found that ID/IG ratio changed with the different carbon sources, and the graphitization degree increased in the order of Glucose-10C/OMS < FA-10C/OMS < DN-10C/OMS.
|
|
Table 1 Surface properties and relative ratio of Co2+/Co in Co/x-10C/OMS catalysts. |

The zeta-potential is a physical property to evaluate the electrical properties of the interfacial layer between materials and the dispersion agent, which could reflect the functionalization degree of carbon materials [30]. As presented in Table 1, the surface of x-10C/OMS supports is negatively charged, which is due to the presence of the electronegative functional groups [31]. It is reported that the dissociation of surface acidic groups (COOH → COO- + H+) would result in a negative zeta potential value. And zeta potential value is related to the amount of surface functional groups [27]. Therefore, the amount of surface functional groups of different samples follows the order of Glucose-10C/OMS > FA-10C/OMS > DN-10C/OMS.
To further semi-quantitative analysis of the surface carbon species, XPS was conducted as well [32]. The C 1s spectra of x-10C/OMS supports are shown in Fig. S4 (Supporting information). The peaks located at 284.5 eV and 285.3 eV are assigned to aromatic carbon (sp2 carbon) and aliphatic carbon (sp3 carbon), respectively [33]. In addition to the C-C groups, the characteristic peaks of oxygenic functional groups also exhibit in the C 1s spectra. The peaks at 286.3 eV, 288.0 eV and 289.0 eV belong to C-O (hydroxyl), C = O (ketone or carboxyl) and O = C-O (carboxyl or esters), respectively [34]. The relative ratio of different functional groups calculated from the deconvolution and quantification of the C 1s spectra are given in Table S2 (Supporting information). Combined with the zeta-potential characterization, it is demonstrated that the amount of surface oxygenic functional groups follows the order of Glucose-10C/OMS > FA-10C/OMS > DN-10C/OMS supports.
It is noteworthy that the amount of functional groups show the same changing tendency with the graphitization degree obtained from Raman spectra. As reported that both the surface functional groups and defects can serve as anchor points of metal sites [35]. Thus, based on the overall analysis of the x-10C/OMS, it is indicated that the carbon source greatly influenced the surface properties, which may further influence the dispersion and catalytic behavior of metal sites.
After successfully fabricated the carbon-layer coated OMS composite, cobalt species was loaded to the support using incipient wetness impregnation method. The cobalt loadings of Co/x-10C/OMS catalysts were determined by ICP-OES. As shown inTable S1, all catalysts have similar cobalt contents, which are close to the designed value of 12 wt%. In order to identify the morphology of the carbonized Co/x-10C/OMS catalysts, TEM images were recorded and shown in Fig. 2. The highly ordered mesostructured can be observed in all samples, and most nanoparticles are encapsulated inside the channels. Compared to Co/OMS catalyst, the carbon layer coating promotes a more uniform distribution of the nanoparticles. As shown in Fig. 2 and Table S1, with a 10 wt% carbon layer-coated structure, the particle size were decreased from 8.6 nm to about 5 nm. In addition, the grain size of Co/x-10C/OMS catalysts derived from different sources increased in the order of Co/Glucose-10C/OMS < Co/FA-10C/OMS < Co/DN-10C/OMS. A lot of researches have reported that the surface defects andfunctional groups can act as the nucleation sites for metal oxide crystallization, which leads to a higher dispersion of active sites [36, 37]. Combined with the above analysis of surface properties of x-10C/OMS supports, we can conclude that carbon coating effectively facilitate the dispersion of Co species. The higher amount of surface functional groups would lead to a smaller particle size.

|
Download:
|
| Fig. 2. TEM images and particle size distribution of carbonized (A) Co/OMS, (B) Co/Glucose-10C/OMS, (C) Co/FA-10C/OMS, and (D) Co/DN-10C/OMS catalysts. | |
XRD patterns of carbonized Co/x-10C/OMS catalysts were conducted to analyze the crystalline structure of cobalt species. As given in Fig. 3, the broad peak centered at 22° belongs to amorphous SiO2. The diffraction peaks at 44.2°, 51.5° and 75.8° are assigned to metallic Co and the peaks at 41.2°, 42.7° and 45.7° correspond to Co2C phase. It can be observed that metallic Co and Co2C exist simultaneously in the carbonized Co/OMS catalyst. But with the carbon layer-coated support, the intensity of diffraction peaks ascribed to Co2C increased distinctly, implying that the carbon coating greatly promoted the formation of Co2C species. There is no obvious existence of the peaks of metallic Co in carbonized Co/x-10C/OMS catalysts, which may be resulted from the high dispersion of metallic Co species. Additionally, the catalysts with carbon layer-coating derived from different sources have no remarkable difference on the distribution of metallic Co and Co2C species.

|
Download:
|
| Fig. 3. XRD patterns (A) and detailed step scanning patterns (B) of carbonized Co/OMS and Co/x-10C/OMS catalysts. | |
XPS was used to further identify the surface states of carbonized Co/x-10C/OMS catalysts. The Co 2p XPS (Fig. S5 in Supporting information) spectra show two peaks at around 780 and 795 eV, which are typically attributed to the binding energy of Co 2p3/2 and Co 2p1/2 respectively. In comparison with Co/DN-10C/OMS, the Co 2p3/2 peaks of Co/FA-10C/OMS and Co/Glucose-10C/OMS shifts to lower binding energies, and the shifting trend is consistent with the amount of surface functional groups. This is due to the electron donor effect of surface oxygenic functional groups on cobalt species [38] Moreover, assuming that Co0 atoms and Co2+ ions have the same surface area and identical atomic sensitivity factors, the surface content ratio of Co2+ to Co can be estimated by calculating the ratio of corresponding fitted peak areas. The binding energy around 778.2 and 793.3 eV are attributed to metallic Co and those at about 782.0 and 798.0 eV could be assigned to Co2+. The appearance of two satellite peaks at 787.5 and 803.7 eV further confirms the existence of Co2+ species. As listed in Table 1, the surface ratio of Co2+ to Co0 remarkably increases from 1.36 to about 2.2 after carbon layer coating. However, there is no significant change in the ratio of Co2+ to Co when the carbon source is different.
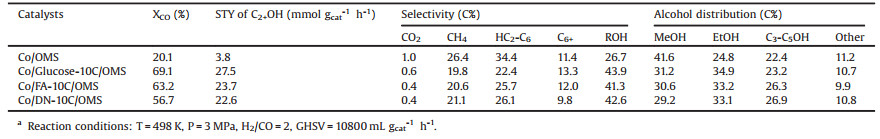
The catalytic performance of Co/OMS and Co/x-10C/OMS catalysts were shown in Table 2. There is only a small number of CO2 produced, which is attributed to the low water-gas shift (WGS) activity of cobalt-based catalysts [39]. Notably, Co/x-10C/OMS catalysts exhibited excellent performance under the GHSV as high as 10800 mL gcat-1 h-1 and relatively low reaction temperature (225 ℃). The carbon layer-coated supports significantly improved the catalytic activity, which could be attributed to the well-dispersed active sites as demonstrated by XRD and TEM results. In addition, the CO conversions of Co/x-10C/OMS catalysts coated by different carbon sources changed following the order of Glucose > FA > DN, which is consistent with the amounts of surface anchor sites. Given the characterization results of Co/x-10C/OMS, the surface functional groups and defects play a key role in promoting the dispersion of active sites, thereby enhancing the catalytic activity.
|
|
Table 2 Catalytic performance of higher alcohol synthesis over Co/OMS and Co/x-10C/OMS catalysts.a |
{kind=link}
{kind=link}
{kind=link}
Moreover, with the carbon layer coating of Co/OMS catalyst, the alcohols selectivity increases from 26.7% to about 40%. The alcohols selectivity among Co/x-10C/OMS catalysts coated by different carbon sources are similar. According to the results of XRD and XPS, carbon coating promotes the formation of Co2+, but different carbon sources have no remarkable influence on the distribution of cobalt species. As reported by Wen et al. [40], the negative charge state of iron particles would strongly weaken the CO bond and promote the carbonization of iron species. In this case, we deduce that carbon layer acts as an electron donor to promote CO disassociation on cobalt species, thereby facilitating the formation of cobalt carbide. A lot of literatures have proposed that the formation of Co2+ species is the key to enhance the alcohols selectivity. They pointed out that Co2+ sites facilitated the CO nondissociative adsorption and molecularly insertion, which was responsible for HAS synthesis [12, 18]. Based on the above investigations, it is demonstrated that the carbon coating of OMS significantly increased the relative ratio of Co2+ to Co0, which further enhanced the alcohols selectivity. The carbon source for coating influenced the metal dispersion, but it is not the main factor affecting the distribution of Co0-Co2+ species.
In summary, a series of carbon layer-coated OMS composites using glucose, FA and DN as precursors were prepared and used as supports for cobalt-based catalysts. The maximum C2+OH space time yield (27.5 mmol gcat-1 h-1) was obtained over Co/Glucose-10C/OMS catalyst. It is found that the carbon layer-coated supports could significantly enhance the metal dispersion and increase the ratio of Co2+ to Co0, which further enhance the CO conversion and alcohols selectivity in HAS. Moreover, the surface properties of x-10C/OMS can be tuned by using different carbon sources. It is demonstrated that the defects and functional groups could effectively disperse cobalt species. The catalytic performance follows the order of Co/Glucose-10C/OMS > Co/FA-10C/OMS > Co/DN-10C/OMS, which is consistent with the amount of surface anchor sites. But the carbon source is not the main factor affecting the distribution of Co-Co2+ dual sites, thus has little effect on the alcohols selectivity. These results may provide new guidance in the rational design of carbon supported catalysts for HAS.
AcknowledgmentsWe gratefully acknowledge support from the National Natural Science Foundation of China (Nos. U1462204, 21706184) and the National Postdoctoral Program for Innovative Talents of China (No. BX20180221).
Appendix A. Supplementary dataSupplementary material related to this article can befound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.04.070.
| [1] |
L. Siwale, L. Kristóf, A. Bereczky, M. Mbarawa, A. Kolesnikov, Fuel Process. Technol. 118 (2014) 318-326. DOI:10.1016/j.fuproc.2013.10.007 |
| [2] |
Q. Mei, X. Shen, H. Liu, B. Han, Chin. Chem. Lett. 30 (2019) 15-24. DOI:10.1016/j.cclet.2018.04.032 |
| [3] |
G.A. Mills, Fuel 73 (1994) 1243-1279. DOI:10.1016/0016-2361(94)90301-8 |
| [4] |
L. Zhong, F. Yu, Y. An, et al., Nature 538 (2016) 84. DOI:10.1038/nature19786 |
| [5] |
N.D. Subramanian, G. Balaji, C. Kumar, J.J. Spivey, Catal. Today 147 (2009) 100-106. DOI:10.1016/j.cattod.2009.02.027 |
| [6] |
R.G. Herman, Catal. Today 55 (2000) 233-245. DOI:10.1016/S0920-5861(99)00246-1 |
| [7] |
J. Su, Z. Zhang, D. Fu, et al., J. Catal. 336 (2016) 94-106. DOI:10.1016/j.jcat.2016.01.015 |
| [8] |
S. Carenco, A. Tuxen, M. Chintapalli, et al., J. Phys. Chem. C 117 (2013) 6259-6266. DOI:10.1021/jp4000297 |
| [9] |
Y. Yang, X. Qi, X. Wang, et al., Catal. Today 270 (2016) 101-107. DOI:10.1016/j.cattod.2015.06.014 |
| [10] |
W. Zi, G. Laddha, S. Kanitkar, J.J. Spivey, Catal. Today 298 (2017) 209-215. DOI:10.1016/j.cattod.2017.04.035 |
| [11] |
X. Song, Y. Ding, W. Chen, et al., Energ. Fuel 26 (2012) 6559-6566. DOI:10.1021/ef301391f |
| [12] |
T. Chen, J. Su, Z. Zhang, et al., ACS Catal. 8 (2018) 8606-8617. DOI:10.1021/acscatal.8b00453 |
| [13] |
H.N. Pham, A.E. Anderson, R.L. Johnson, et al., ACS Catal. 5 (2015) 4546-4555. DOI:10.1021/acscatal.5b00329 |
| [14] |
H. Xiong, Y. Zhang, K. Liew, J. Li, J. Mol. Catal. A 295 (2008) 68-76. DOI:10.1016/j.molcata.2008.08.017 |
| [15] |
V.N. Mochalin, O. Shenderova, D. Ho, Y. Gogotsi, Nat. Nanotechnol. 7 (2012) 11-23. DOI:10.1038/nnano.2011.209 |
| [16] |
Y. Chen, G. Zhao, L. Lin, Chin. Chem. Lett. 29 (2018) 1633-1636. DOI:10.1016/j.cclet.2018.01.055 |
| [17] |
X. Dong, X.L. Liang, H.Y. Li, et al., Catal. Today 147 (2009) 158-165. DOI:10.1016/j.cattod.2008.11.025 |
| [18] |
Y.P. Pei, J.X. Liu, Y.H. Zhao, et al., ACS Catal. 5 (2015) 3620-3624. DOI:10.1021/acscatal.5b00791 |
| [19] |
D. Zhao, J. Feng, Q. Huo, et al., Science 279 (1998) 548-552. DOI:10.1126/science.279.5350.548 |
| [20] |
L. Dai, W. Bo, F. Tan, et al., Bioresource Technol. 161 (2014) 327-332. DOI:10.1016/j.biortech.2014.03.086 |
| [21] |
R. Schmidt, E.W. Hansen, M. Stoecker, D. Akporiaye, O.H. Ellestad, J. Am. Chem. Soc. 117 (1995) 4049-4056. DOI:10.1021/ja00119a021 |
| [22] |
D.S.T. Martinez, J.P.V. Damasceno, L.S. Franqui, et al., Mater.Sci. Eng.C 78 (2017) 141-150. DOI:10.1016/j.msec.2017.03.298 |
| [23] |
H. Nishihara, Y. Fukura, K. Inde, et al., Carbon 46 (2008) 48-53. DOI:10.1016/j.carbon.2007.10.024 |
| [24] |
H. Nishihara, T. Kwon, Y. Fukura, et al., Chem. Mater. 23 (2011) 3144-3151. DOI:10.1021/cm103388y |
| [25] |
X.Y. Liu, L.B. Sun, X.D. Liu, et al., ACS Appl. Mater. Inter. 5 (2013) 9823-9829. DOI:10.1021/am403033z |
| [26] |
K. Krishnamoorthy, U. Navaneethaiyer, R. Mohan, J. Lee, S.J. Kim, Appl. Nanosci. 2 (2012) 119-126. DOI:10.1007/s13204-011-0045-9 |
| [27] |
K. Krishnamoorthy, M. Veerapandian, K. Yun, S.J. Kim, Carbon 53 (2013) 38-49. DOI:10.1016/j.carbon.2012.10.013 |
| [28] |
F. Coloma, A. Sepulvedaescribano, F. Rodriguezreinoso, J. Catal. 154 (1995) 299-305. DOI:10.1006/jcat.1995.1171 |
| [29] |
A.C. Ferrari, J. Robertson, Phys. Rev. B 61 (2000) 14095-14107. DOI:10.1103/PhysRevB.61.14095 |
| [30] |
T.Y. Kim, H.W. Lee, M. Stoller, et al., ACS Nano 5 (2011) 436-442. DOI:10.1021/nn101968p |
| [31] |
I. Jung, D.A. Dikin, R.D. Piner, R.S. Ruoff, Nano Lett. 8 (2008) 4283-4287. DOI:10.1021/nl8019938 |
| [32] |
D.M. Hercules, J. Electron Spectrosc. 5 (1974) 811-826. DOI:10.1016/0368-2048(74)85054-1 |
| [33] |
A. Ambrosi, M. Pumera, Chem.-Eur. J. 19 (2013) 4748-4753. DOI:10.1002/chem.201204226 |
| [34] |
S. Reiche, R. Blume, X.C. Zhao, et al., Carbon 77 (2014) 175-183. DOI:10.1016/j.carbon.2014.05.019 |
| [35] |
P. Serp, B. Machado, Nanostructured Carbon Materials for Catalysis, (Catalysis Series), Royal Society of Chemistry, London, 2015.
|
| [36] |
B.F. Machado, O. Mustapha, A.M. Rosa, et al., J. Catal. 309 (2014) 185-198. DOI:10.1016/j.jcat.2013.09.016 |
| [37] |
C. Liu, Y. He, L. Wei, et al., ACS Catal. 8 (2018) 1591-1600. DOI:10.1021/acscatal.7b03887 |
| [38] |
M. Franz, H.A. Arafat, N.G. Pinto, Carbon 38 (2000) 1807-1819. DOI:10.1016/S0008-6223(00)00012-9 |
| [39] |
J.C. Mohandas, M.K. Gnanamani, G. Jacobs, et al., ACS Catal. 1 (2011) 1581-1588. DOI:10.1021/cs200236q |
| [40] |
X. Zhou, G.J.A. Mannie, J. Yin, et al., ACS Catal. 8 (2018) 7326-7333. DOI:10.1021/acscatal.8b02076 |