 2020, Vol. 31
2020, Vol. 31 


b Limin Chemical Co., Ltd., Xinyi 221422, China;
c Beijing Key Laboratory of Drug Resistance Tuberculosis Research, Department of Pharmacology, Beijing Tuberculosis and Thoracic Tumor Research Institute, Beijing Chest Hospital, Capital Medical University, Beijing 100149, China
Tuberculosis (TB), which is mainly caused by Mycobacterium tuberculosis (MTB), remains one of the world's deadliest pandemic diseases with approximately 10.0 million new TB cases and 1.6 million TB-related deaths estimated by the WHO in 2017 [1]. The sprawl of multidrug-resistant (MDR)-TB and extensively drugresistant (XDR) TB have created an urgent demand for the discovery and development of novel TB drugs to improve the treatment outcomes [2-4]. Encouragingly, bedaquiline and delamanid have been approved for the treatment of MDR-TB, although some adverse events have been noted [5]. Considering the high attrition rate in new drug development, there is still a great need for safer and more effective candidates with novel mechanisms of action [6].
Imidazo[1, 2-a]pyridine-3-carboxamides (IPAs) as new anti-TB agents targeting QcrB [7] have garnered great interest recently. The candidate Q203 (Fig. 1) is in phase Ⅱ clinical trials at present [1], and many series of new IPAs were reported to have potent antimycobacterial activity [8-19]. Structure-activity relationship (SAR) studies of IPAs demonstrate that the carboxamide linker with the N-benzylic group is critical for anti-MTB activity [16]. In our previous studies [17-19], however, some N-(2-phenoxy)ethyl IPAs exampled by WZY02 (Fig. 1), have been identified as new anti-TB agents with strong in vitro anti-MTB activity. Herein we report on the additional SAR studies of 2, 6-disubstituted N-(2-phenoxy)ethyl IPAs containing various amine moieties. Our primary objective was to find new IPA derivatives with potent anti-MTB activity and facilitate the further development of these compounds.

|
Download:
|
| Fig. 1. Structures of Q203 and WZY02. | |
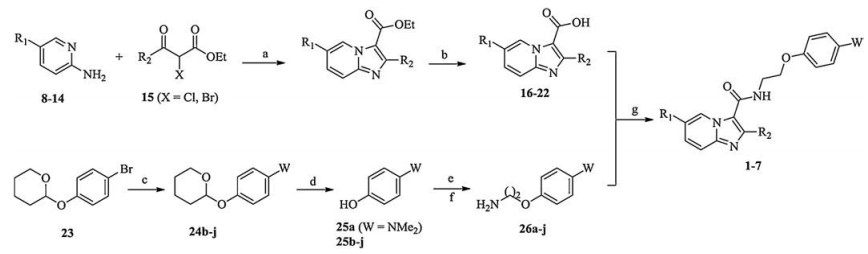
The synthesis of target compounds 1–7 are shown in Scheme 1. As our previous procedures [20], IPA core acids 16–22 were easily obtained via microwave assistant cyclization of pyridine-amines 8–14 and esters 15, and then hydrolysis. Buchwald-Hartwig amination of THP (tetrahydro-2H-pyran)-protected bromophenol 23 with amines gave condensates 24b–j, and removal of the THP group of 24b–j yielded para-aminophenols 25b–j. Nucleophilic substitution of 25b–j and commercially available 25a with bromoacetonitrile, and then reduction gave 4-(2-aminoethoxy)-anilines 26a–j. Coupling of acids 16–22 with side-chain compounds 26a–j achieved IPA derivatives 1–7.

|
Download:
|
| Scheme 1. Synthesis of 1–7. Reagents and conditions: (a) MW, EtOH, 120 ℃; (b) LiOH, THF, H2O, 0–5 ℃; (c) (±)-2, 2'-Bis(diphenylphosphino)-1, 1'-binaphalene, Cs2CO3, palladium(Ⅱ) acetate, amines, toluene, reflux; (d) EtOH, 4-methylbenzenesulfonate, pyridin-1-ium; reflux; (e) K2CO3, bromoacetonitrile, DMSO, r.t.; (f) LiAlH4, THF, r.t.; (g) BOP-Cl, Et3N, DCM, r.t. | |
The target compounds 1–7 were initially screened for in vitro activity against MTB H37Rv ATCC 27294 strain using the Microplate Alamar Blue Assay (MABA) [21, 22]. The minimum inhibitory concentration (MIC) is defined as the lowest concentration effecting a reduction in fluorescence of >90% relative to the mean of replicate bacterium-only controls. The MIC values of the compounds along with isoniazid (INH), rifampicin (RFP), Q203, PBTZ169 and the lead compound WZY02 for comparison are presented in Table 1. Detailed procedures are shown in Supporting information. The data reveal that most of them have good potency against this strain (MIC: < 0.05 μg/mL). Twenty compounds were found to show the same excellent activity (MIC: < 0.016 μg/mL) as Q203 and PBTZ169, while being more active than INH, RFP and WZY02 (MIC: 0.031-0.059 μg/mL).
|
|
Table 1 Structures and activity of compounds 1–7 against MTB H37Rv. |
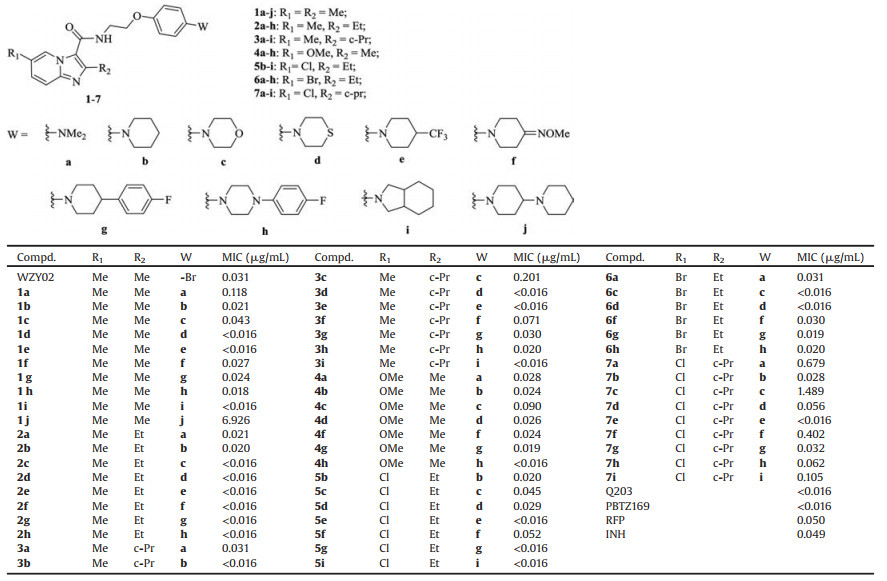
We first investigated the potential impact of various amines on the anti-MTB activity of the 2, 6-dimethyl IPA series. Replacement of the bromine atom on the para-position of the benzene ring of the lead WZY02 (MIC: 0.031 μg/mL) with dimethylamine group in compound 1a leads to decreased activity (MIC: 0.118 μg/mL), but the presence of piperidine in compound 1b demonstrates better potent MIC value of 0.021 μg/mL, suggesting that cyclic amine is preferred over non-cyclic one. And this phenomenon is also observed in compounds 1c and 1d containing morpholine and thiomorpholine rings, the isostere of the piperidine (1b), respectively.
Further investigation reveals that introduction of an electronwithdrawing group, a methyloxime (a known functional moiety of the third and fourth generational cephalosporins) or an aromatic moiety on the para-position of the piperidine ring is acceptable, such as compounds 1e–g with MIC of < 0.016–0.027 μg/mL. As expected, the piperidine ring of 1g could be replaced by its isostere piperazine (1h, MIC: 0.018 μg/mL) without obviously affecting the anti-MTB potency. Interestingly, compound 1i with a fused nitrogen heterocycle (octahydro-1H-isoindole) shows the best activity (MIC: < 0.016 μg/mL). However, introduction of an additional piperidyl group on the para-position of the piperidine ring of 1b is detrimental, and the resulting compound 1j shows poor activity (MIC: 6.926 μg/mL) probably due to the alkaline of the terminal piperidine ring, indicating that 1, 4'-bipiperidine is unacceptable as the W group. The above results suggest that all the amines with an exception of 1, 4'-bipiperidine (j), could be ideally suited as the W groups.
Subsequently, the effect of the R1 and R2 groups on the anti-MTB activity of other six 2, 6-disubstituted IPA series with nine amine moieties (a–i) was further investigated. With a few exceptions, as shown in Table 1, replacement of the methyl group (R2) on the core of 2, 6-dimethyl IPA series 1a–i with ethyl (2a–i) results to slightly increased potency, but cyclopropyl (c-Pr) as R2 (3a–i) has not obvious influence on the activity, indicating that ethyl group as R2 is preferred over methyl and cyclopropyl. Moreover, the C-6 position of the IPA core is also well tolerated by another electrondonating OMe group (1a–i vs. 4a–i).
In further modifications, the methyl group of series 2 or 3 was replaced by a halogen atom as the R1 group. 6-Chloro-2-ethyl IPA derivatives 5 with the same core of Q203, As shown in Table 1, display roughly the same activity (MIC: < 0.016–0.052 μg/mL) as the corresponding analogues 2. Replacement of the chlorine atom of series 5 with bromine does not affect significantly the activity (5 vs. 6). But the presence of cyclopropyl group instead of the ethyl as the R2 group (5 vs. 7), or chlorine instead of the methyl as the R1 group (3 vs. 7) seems to be unfavorable. Overall, the above results indicate that anti-MTB activity of the N-(2-phenoxy)ethyl IPA derivatives is related to R1 and R2 groups of the IPA core as well as W on the para-position of the benzene ring.
Encouraged by their potent activity (MIC: < 0.016 μg/mL) against drug sensitive MTB H37Rv strain, 20 compounds were evaluated against two clinical isolated MTB-MDR (16995 and 16833) strains resistant to both INH and RFP. As shown in Table 2, all of them exhibit strong potency (MIC: < 0.002–0.030 μg/mL), especially 13 compounds have the same best MIC values of < 0.002 μg/mL as Q203 and PBTZ169, suggesting their promising potential for both drug-sensitive and resistant MTB strains (Tables 1 and 2).
|
|
Table 2 Anti-MDR-MTB activity and cytotoxicity of selected compounds. |
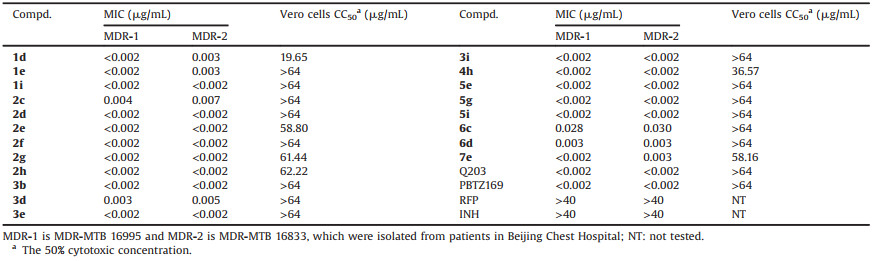
The above 20 compounds were also tested for mammalian cell cytotoxicity using Vero cells measured as a concentration inhibiting 50% growth (CC50) as compared to a no-treatment control [22]. As shown in Table 2, 14 compounds show low cytotoxicity (CC50: > 64 μg/mL) comparable to Q203 and PBTZ169. Detailed procedures are shown in Supporting information.
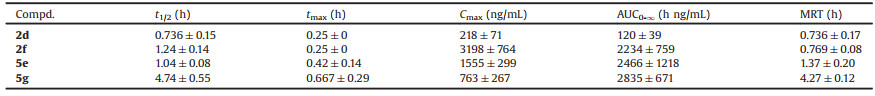
Based on the measured activity against all the tested strains and cell cytotoxicity, 4 compounds 2d, 2f, 5e and 5h were further evaluated for their in vivo pharmacokinetic (PK) profiles in ICR female mice after a single oral administration of 25 mg/kg. Compared with 2d, as shown in Table 3, methyloxime-containing compound 2f with the same 2-ethyl-6-methyl IPA core, displays significantly higher Cmax (3198 ng/mL), AUC0-∞ (2234 h ng/mL) and longer t1/2 (1.24 h). Although compound 5e shows acceptable PK profiles, its analogue 5g with 4-fluorophenyl instead of trifluoromethyl group on the para-position of the piperidine ring demonstrates higher AUC0-∞ (2835 h ng/mL) and longer t1/2 (4.74 h). Detailed procedures are shown in Supporting information.
|
|
Table 3 In vivo PK profiles of selected compounds dosed orally in ICR female mice at 25 mg/kg (n = 3). |
{kind=link}
{kind=link}
In conclusion, seven 2, 6-disubstituted N-(2-phenoxy)ethyl IPA series containing various amine moieties were designed and synthesized as new anti-TB agents. Many of them exhibit excellent in vitro inhibitory activity with low nanomolar MIC values against both drug-sensitive MTB strain H37Rv and two drug-resistant clinical isolates (MIC: < 0.002–0.030 μg/mL), and low cytotoxicity (CC50: > 64 μg/mL). Particularly, compounds 2f, 5e and 5g show acceptable PK profiles, suggesting they may serve as new and promising candidates for further study.
AcknowledgmentsThis work is supported by the CAMS Innovation Fund for Medical Science (No. CAMS-2017-I2M-1-011), National Mega-project for Innovative Drugs (Nos. 2015ZX09102007-008, 2018ZX09721001-004-007, 2018ZX09711001-007-002), the National Natural Science Foundation of China (No. 81872753).
Appendix A. Supplementary dataSupplementary material related to this article can befound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.07.038.
| [1] |
World Health Organization, Global Tuberculosis Report, 2018. http://www.who.int/tb/.
|
| [2] |
Z. Xu, S. Zhang, C. Gao, et al., Chin. Chem. Lett. 28 (2017) 159-167. DOI:10.1016/j.cclet.2016.07.032 |
| [3] |
Y. Zhang, R. Wang, T. Zhang, et al., Chin. Chem. Lett. 30 (2019) 653-655. DOI:10.1016/j.cclet.2018.11.032 |
| [4] |
A. Wang, Y. Yang, Y. Jun, et al., Bioorg. Med. Chem. 26 (2018) 2073-2084. DOI:10.1016/j.bmc.2018.03.004 |
| [5] |
H Wang, K. Lv, X. Li, et al., Chin. Chem. Lett. 30 (2019) 413-416. DOI:10.1016/j.cclet.2018.08.005 |
| [6] |
S. Tiberi, R. Buchanan, J.A. Caminero, et al., Presse Med. 46 (2017) e41-e51. DOI:10.1016/j.lpm.2017.01.016 |
| [7] |
K.A. Abrahams, J.A. Cox, V.L. Spivey, et al., PloS One 7 (2012) e52951. DOI:10.1371/journal.pone.0052951 |
| [8] |
G.C. Moraski, L.D. Markley, J. Cramer, et al., ACS Med. Chem. Lett. 4 (2013) 675-679. DOI:10.1021/ml400088y |
| [9] |
G.C. Moraski, L.D. Markley, P.A. Hipskind, et al., ACS Med. Chem. Lett. 2 (2011) 466-470. DOI:10.1021/ml200036r |
| [10] |
G.C. Moraski, L.D. Markley, M. Chang, et al., Bioorg. Med. Chem. 7 (2012) 2214-2220. DOI:10.1016/j.bmc.2012.02.025 |
| [11] |
H. Wang, A. Wang, J. Gu, et al., Eur. J. Med. Chem. 165 (2019) 11-17. DOI:10.1016/j.ejmech.2018.12.071 |
| [12] |
A. Wang, H. Wang, Y. Geng, et al., Bioorg. Med. Chem. 27 (2019) 813-821. DOI:10.1016/j.bmc.2019.01.022 |
| [13] |
S. Kang, Y.M. Kim, R.Y. Kim, et al., Eur. J. Med. Chem. 125 (2017) 807-815. DOI:10.1016/j.ejmech.2016.09.082 |
| [14] |
S. Kang, Y.M. Kim, H. Jeon, et al., Eur. J. Med. Chem. 136 (2017) 420-427. DOI:10.1016/j.ejmech.2017.05.021 |
| [15] |
G.C. Moraski, L.D. Markley, P.A. Hipskind, et al., ACS Med. Chem. Lett. 2 (2011) 466-470. DOI:10.1021/ml200036r |
| [16] |
S. Kang, R.Y. Kim, M.J. Seo, et al., J. Med. Chem. 57 (2014) 5293-5305. DOI:10.1021/jm5003606 |
| [17] |
K. Lv, L. Li, B. Wang, et al., Eur. J. Med. Chem. 137 (2017) 117-125. DOI:10.1016/j.ejmech.2017.05.044 |
| [18] |
Z. Wu, Y. Lu, L. Li, et al., ACS Med. Chem. Lett. 7 (2016) 1130-1133. DOI:10.1021/acsmedchemlett.6b00330 |
| [19] |
A. Wang, K. Lv, L. Li, et al., Eur. J. Med. Chem. 175 (2019) 715-725. |
| [20] |
L. Li, Z. Wu, M. Liu, et al., Heterocycles 91 (2015) 2087-2095. DOI:10.3987/COM-15-13299 |
| [21] |
L.A. Collins, S.G. Franzblau, Antimicrob. Agents Chemother. 41 (1997) 1004-1009. DOI:10.1128/AAC.41.5.1004 |
| [22] |
Y. Lu, M. Zheng, B. Wang, et al., Antimicrob. Agents Chemother. 55 (2011) 5185-5193. DOI:10.1128/AAC.00699-11 |