 2020, Vol. 31
2020, Vol. 31 


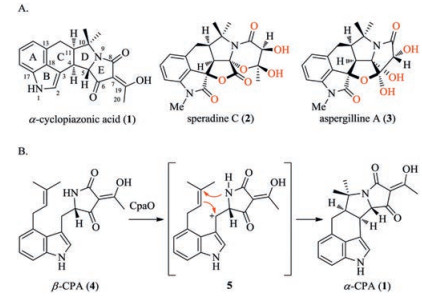
α-Cyclopiazonic acid (α-CPA, 1), a mycotoxin, was firstly isolated from the fungus Penicillium cyclopium Westling, which is often isolated from stored grain and cereal products (Fig. 1A) [1]. In addition, α-CPA is also a nanomolar inhibitor of sarco/endoplasmic reticulum Ca2+-ATPase (SERCA), which are essential for calcium reuptake in muscle contraction and relaxation cycles [2-4]. Importantly, SERCA is a promising target for the development of new drugs against various diseases and insect pests. Thus, α-CPA is one of the few potent, selective and reversible SERCA inhibitors which could act as a lead in drug development.

|
Download:
|
| Fig. 1. (A) Structures of α-cyclopiazonic acid, speradines C, and aspergilline A. (B) Biosynthesis of α-CPA. | |
{kind=link}
Structurally, α-CPA is a distinguished 3, 4-fused indole alkaloid containing a 6/5/6/5/5 pentacyclic ring system and a highly substituted tetramic acid moiety (Fig. 1A) [1]. Since 2003, a number of highly oxygenated α-CPA-derived alkaloids, such as speradines A–C and aspergillines A–E, were isolated and identified [5-10].
Biosynthetic studies have revealed that β-cyclopiazonic acid (β-CPA, 4) is the direct biosynthetic precursor of α-CPA (Fig. 1B) [11, 12]. Oxidation of 4 by the flavin-dependent monoamine oxidase Mao A (CpaO) to generate a benzylic cation 5 that subsequently undergoes cascade cyclization to result in formation of the C/D rings of 1.
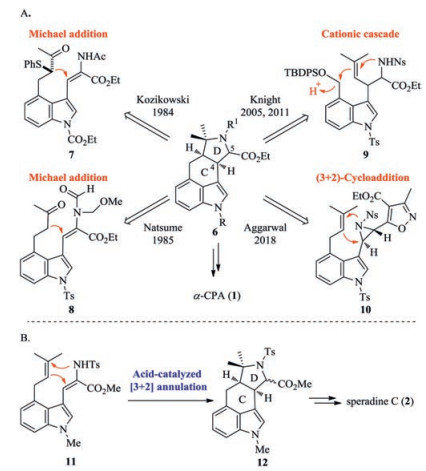
To date, five total syntheses of α-CPA have been reported (Scheme 1A) [13-19]. The Kozikowski and Natsume groups constructed the C and D rings in a stepwise manner in which the C ring was formed by an intramolecular Michael addition [13, 14]. In 2005, the Knight group assembled the C/D rings with high stereocontrol by an elegant carbocationic cascade [15, 16]. In 2011, the Scherkenbeck group accomplished the first asymmetric synthesis of 1 by a modification of the Knight synthesis and found that cyclization of the same substrate 9 produced a 1:1 mixture of diastereomers across the C/D ring junction [17, 18]. In 2018, the Aggarwal group reported the enantioselective synthesis of 1 by a formal [3 + 2] cycloaddition to form the C/D rings [19].

|
Download:
|
| Scheme 1. (A) Previous syntheses of α-CPA. (B) Our strategy for the synthesis of speradine C (2). | |
{kind=link}
As part of our ongoing studies towards the concise and efficient synthesis of 3, 4-indole alkaloids [20-25], we are attracted by the structure and biological activity of α-CPA and the related natural products. Inspired by the biosynthesis of α-CPA (Fig. 1B), we have discovered an unprecedented acid-catalyzed [3 + 2] annulation of N-Me dehydrotryptophan derivative 11 to produce tetracycle 12 by formation of the C/D rings, which enabled a ten-step total synthesis of speradine C (2) (Scheme 1B) [26]. Encouraged by this result, we envisioned that the cascade cyclization of the analogous N-Ts dehydrotryptophan derivative could be applied to the total synthesis of α-CPA. Herein, we report a concise total synthesis of α-CPA by using this novel acid-catalyzed [3 + 2] annulation.
Retrosynthetic analysis of α-CPA (1) was shown in Scheme 2. We envisioned that the tetramic acid residue of 1 could be generated from tetracycle 13 and diketene 14 by Dieckmann cyclization (Scheme 2) [14]. The C/D rings of the key tetracycle 13 could be constructed by acid-catalyzed [3 + 2] annulation of the indole N-Ts dehydrotryptophan derivative 15. In turn, the cyclization precursor 15 could be readily prepared from compounds 16-18.

|
Download:
|
| Scheme 2. Retrosynthetic analysis of α-CPA. | |
{kind=link}
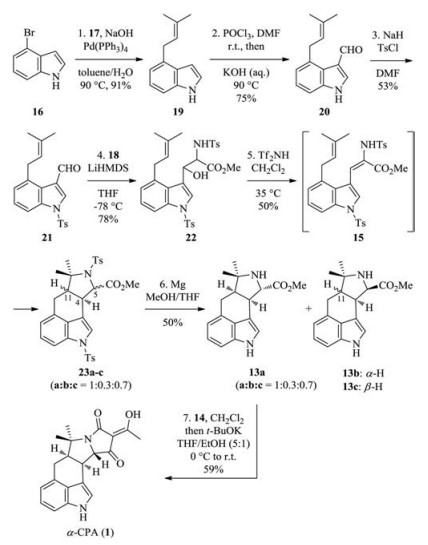
Our synthesis commenced with 4-bromoindole 16, which underwent a Suzuki cross-coupling with tert-prenylboronate 17 to give indole 19 as the sole regioisomer (Scheme 3) [27]. VilsmeierHaack formylation of 19 followed by N-tosylation provided the desired aldehyde 21. Aldol reactions of N-Ts amino acid esters 18 with aldehyde 21 yielded the corresponding α-amino β-hydroxy ester 22 as a mixture of syn/anti diastereoisomers (dr = 1:1), which could be separated and identified by careful column chromatography. Considering that compound 22 would then be converted to dehydrotryptophan derivative 15 that has no chiral center, compound 22 could be directly used for next step without separation.

|
Download:
|
| Scheme 3. Total synthesis of α-CPA. | |
{kind=link}
With the α-amino β-hydroxy ester 22 in hand, we then investigated the critical [3 + 2] annulation. To our delight, compound 22 could be readily converted to the desired tetracycle 23a–c (dr = 1:0.3:0.7 at C-5 and C-11) through dehydrotryptophan derivative 15 under the optimal reaction conditions (0.2 equiv. of Tf2NH in CH2Cl2 at 35 ℃) [26]. Tetracycles 23a–c were very hard to be separated by column chromatography, and only small amount of pure 23a was obtained.
Having successfully constructed the C/D ring system, we turned our attention to the synthesis of α-CPA (1). Atthis stage, weneeded to remove the two Ts groups in 23, however, it proved exceptionally difficult, especially for the pyrrolidine N-Ts group. After several trials, removal of the two Ts groups in 23a–c with a large excess of amount of Mg in MeOH/THF gave amines 13a–c, and 13a could be readily separated from 13b and 13c bychromatography. The relative configurations of 13a–c were determined by extensive NMR spectra analysis (the Supporting information) and comparison with the known N-Me-13a–c (dr = 1:0.3:0.7 at C-5 and C-11) [26]. Finally, treatment of tetracycle 13a with diketene 14 in CH2Cl2 followed by stirring with t-BuOK in THF/EtOH (5:1) led smoothly to give α-CPA (1). The physical data of our synthesized α-CPA (1) were identical to those reported in the literature.
In summary, we have achieved the total synthesis of α-CPA in only seven steps from commercially available 4-bromoindole (16). The synthesis features an acid-catalyzed [3 + 2] annulation to form the tetracyclic skeleton. This synthesis represents the shortest pathway for the total synthesis of α-CPA to date.
AcknowledgmentsThis research was supported by the National Natural Science Foundation of China (No. 21871013) and the Drug Innovation Major Project (No. 2018ZX09711-001-005-005).
Appendix A. Supplementary dataSupplementary material related to this article can befound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.06.048.
| [1] |
C.W. Holzapfel, Tetrahedron 24 (1968) 2101-2119. DOI:10.1016/0040-4020(68)88113-X |
| [2] |
R.T. Riley, D.E. Goeger, H. Yoo, J.L. Showker, Toxicol. Appl. Pharmacol. 114 (1992) 261-267. DOI:10.1016/0041-008X(92)90076-5 |
| [3] |
F. Martínez-Azorín, FEBS Lett. 576 (2004) 73-76. DOI:10.1016/j.febslet.2004.08.064 |
| [4] |
K. Moncoq, C.A. Trieber, H.S. Young, J. Biol. Chem. 282 (2007) 9748-9757. DOI:10.1074/jbc.M611653200 |
| [5] |
X.H. Ma, J.X. Peng, G.W. Wu, et al., Tetrahedron 71 (2015) 3522-3527. DOI:10.1016/j.tet.2015.03.050 |
| [6] |
M. Tsuda, T. Mugishima, K. Komatsu, et al., Tetrahedron 59 (2003) 3227-3230. DOI:10.1016/S0040-4020(03)00413-7 |
| [7] |
M. Zhou, M.M. Miao, G. Du, et al., Org. Lett. 16 (2014) 5016-5019. DOI:10.1021/ol502307u |
| [8] |
X. Hu, Q.W. Xia, Y.Y. Zhao, et al., Chem. Pharm. Bull. 62 (2014) 942-946. DOI:10.1248/cpb.c14-00312 |
| [9] |
H. Zhu, C. Chen, J. Wang, et al., Chem. Biodivers. 12 (2015) 1547-1553. DOI:10.1002/cbdv.201400412 |
| [10] |
V. Uka, G.G. Moore, N. Arroyo-Manzanares, et al., Toxins 9 (2017) 35-55. DOI:10.3390/toxins9010035 |
| [11] |
P.K. Chang, K.C. Ehrlich, I. Fujii, Toxins 1 (2009) 74-99. DOI:10.3390/toxins1020074 |
| [12] |
X. Liu, C.T. Walsh, Biochemistry 48 (2009) 8746-8757. DOI:10.1021/bi901123r |
| [13] |
A.P. Kozikowski, M.N. Greco, J.P. Springer, J. Am. Chem. Soc. 106 (1984) 6873-6874. DOI:10.1021/ja00334a084 |
| [14] |
H. Muratake, M. Natsume, Heterocycles 23 (1985) 1111-1117. DOI:10.3987/R-1985-05-1111 |
| [15] |
C.M. Haskins, D.W. Knight, Chem. Commun. (2005) 3162-3164. DOI:10.1039/b417625c |
| [16] |
C.M. Griffiths-Jones, D.W. Knight, Tetrahedron 67 (2011) 8515-8528. DOI:10.1016/j.tet.2011.08.094 |
| [17] |
C. Beyer, J. Scherkenbeck, F. Sondermann, A. Figge, Tetrahedron 66 (2010) 7119-7123. DOI:10.1016/j.tet.2010.06.092 |
| [18] |
W.R. Christian Beyer, K. Woithe, B. Lüke, et al., Tetrahedron 67 (2011) 3062-3070. DOI:10.1016/j.tet.2011.03.002 |
| [19] |
O. Zhurakovskyi, Y.E. Turkmen, L.E. Loffler, et al., Angew. Chem. Int. Ed. 57 (2018) 1346-1350. DOI:10.1002/anie.201712065 |
| [20] |
H. Qin, Z.R. Xu, Y.X. Cui, Y.X. Jia, Angew. Chem. Int. Ed. 50 (2011) 4447-4449. DOI:10.1002/anie.201100495 |
| [21] |
D. Shan, Y. Gao, Y.X. Jia, Angew. Chem. Int. Ed. 52 (2013) 4902-4905. DOI:10.1002/anie.201300571 |
| [22] |
L. Li, Q. Yang, Y. Wang, Y.X. Jia, Angew. Chem. Int. Ed. 54 (2015) 6255. DOI:10.1002/anie.201411338 |
| [23] |
H.C. Liu, X.W. Zhang, D. Shan, et al., Org. Lett. 19 (2017) 3323-3326. DOI:10.1021/acs.orglett.7b01504 |
| [24] |
J.B. Lv, B. Wang, K. Yuan, Y. Wang, Y.X. Jia, Org. Lett. 19 (2017) 3664-3667. DOI:10.1021/acs.orglett.7b01681 |
| [25] |
H.C. Liu, Y.X. Jia, Nat. Prod. Rep. 34 (2017) 411-432. DOI:10.1039/C6NP00110F |
| [26] |
H.C. Liu, L.J. Chen, K. Yuan, Y.X. Jia, Angew. Chem. Int. Ed. 58 (2019) 6362-6365. DOI:10.1002/anie.201902004 |
| [27] |
J.W. Clary, T.J. Rettenmaier, R. Snelling, et al., J. Org. Chem. 76 (2011) 9602-9610. DOI:10.1021/jo201093u |