 2020, Vol. 31
2020, Vol. 31 


Recently, more and more attention was paid to CO2 utilizing as green C1 building block in organic synthesis due to its good features such as abundance, nontoxicity and renewability [1-4]. Up to now, several useful products from CO2 as C1 feedstock, e.g., urea, salicylic acid, organic carbonates, methanol and polycarbonates on a large scale have been produced in the chemical industry which consumed approximately 116 million tons of CO2 per year [5].Nevertheless, the chemical varieties are limited which results from the hindrance by the efficiency and economic respects. Therefore, to utilize CO2 in an effective and low-cost manner will be of great significance and urgently required.
Due to the thermodynamic stability and kinetic inertness of CO2 molecule, the activation of CO2 is indispensable by the highly active species in the transformation [6, 7]. In the previous studies, most of the effective catalytic systems still depended on the transition metal catalysis [8, 9]. Most of the active metal complexes were thermolabile and sensitive to air/water. In addition, much attention was paid to the organocatalysis because of its cheap, nontoxic, easily tunable skeleton and multiple functional sites features [10, 11]. It also represented a forceful tool to effectively activate CO2 molecule and promote its conversion under mild reaction conditions. In the reported works, many transformations on CO2 through organocatalysis manner were achieved such as N-heterocyclic carbene [12-15], N-heterocyclic olefin [16, 17], frustrated Lewis pairs [18-21], superbases [22-24], task-specific ionic liquids [25-27] and others [24, 28-30].
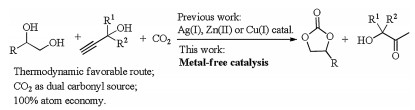
Three-component cascade reaction of propargylic alcohols, CO2 and vicinal alcohols provided a thermodynamically favorable access to cyclic carbonates and α-hydroxyl ketones [31]. In the process, CO2 acted as both carbon and oxygen source for carbonyl group construction with 100% atom economy. Reported systems were based on transition metals such as Ag(I) [31, 32], Cu(I) [33], and Zn(II) [34] (Scheme 1), and the study of organocatalysis on the process has not been involved.

|
Download:
|
| Scheme 1. Thermodynamically favorable cascade reaction for CO2 conversion. | |
Among the organocatalysis, strong base was revealed for its good properties such as easily available material and rich electronegativity which was beneficial for the activation of CO2 [35, 36] and hydroxyl [37, 38] through strong hydrogen bond interaction. On the basis of the theory, we proposed that the amidine base could be the potential catalyst to effectively promote the cascade reaction of propargylic alcohols, CO2 and vicinal alcohols to cyclic carbonates and α-hydroxyl ketones. Delightedly, herein, metal-free catalysis for the three-component reaction was disclosed to produce cyclic carbonates and α-hydroxyl ketones with satisfactory yields of products (Scheme 1).
We thus started to explore the practicability of the proposed conjugated strong base-promoted method in Scheme 1. Initially, a reaction of propylene glycol 1a, propargylic alcohol 2a, and CO2 in CH3CNunder the given conditions was investigated (Table 1). To our delight, when 1, 8-diazabicyclo[5.4.0]undec-7-ene (DBU) was employed as the catalyst, the corresponding products, i.e., propylene carbonate (PC, 3a) and α-hydroxy ketone 4a were obtained in 61% and 58% yields, respectively (entry 1). At the same time, several typical guanidine bases were also examined as catalyst. As seen from the results (entries 2–4), theyalso showed catalytic activity for the transformation. 1, 5-Diazabicyclo[4.3.0]non-5-ene (DBN) possessing the similar structure with DBU gave the equivalent level result (entry 2). The other strong base such as 1, 5, 7-triazabicyclo[4.4.0]dec-5-ene (TBD) and 1, 1, 3, 3-tetramethylguanidine (TMG) gave lower yields (entries 3 and 4). As we all know, the basicity strength of the catalyst plays a key role in the coupling reaction of CO2 and propargylic alcohol, which is the ratedetermining step in the three-component reaction [32, 39]. Generally, the weak basicity of catalysts resulted in low activity, while the strong base exhibited a higher conversion and a lower yield because of some side reactions [38]. The basicity of the guanidine bases (entries 1–4) followed the order: TBD > DBN > DBU > TMG [39-41]. However, their catalytic activity showed as DBU, DBN > TBD > TMG, an action from both basicity and nucleophilicity indicating a good agreement with the previous works [38, 42]. Additionally, both K2CO3 and Cs2CO3 led to the lower yields (entries 5 and 6). The unconjugated base, i.e ., Et3N exhibited significantly a lower catalytic activity than that of guanidine bases (entries 7 vs. 1–4). Clearly, the guanidine bases displayed better performance than that of the other basic catalysts.
|
|
Table 1 The effect of different basic catalysts and solvents on the synthesis of 3a and 4a.a |

Furthermore, the reactions in different solvents were explored in the presence of DBU, and the results revealed the obvious influence of the medium. The use of DMF as the solvent gave a higher yield than that of CH3CN, dimethyl sulfoxide (DMSO) or CH2Cl2 (entries 9 vs. 1, 10 and 11). In addition, the reaction worked with less efficiency under the solvent-free conditions (entry 8).
Next, the effect of CO2 pressure on the reaction was tested, and the results were shown in Fig. 1a. No target product was detected under the atmospheric pressure. However, the yield of product obviously increased when CO2 pressure was from 2 to 5 MPa. Especially, the best result was obtained under 3 MPa, and the yields of 3a and 4a were 79% and 85%, respectively. Notably, due to the formation of carbonic acid from high pressure CO2 and trace water in the reaction system, the activity of the strong base DBU might be weakened [43, 44]. As a result, a slightly lower product yield was obtained in 4MPa and 5 MPa CO2. As shown in Fig. 1b, reaction temperature had a great influence on the process. 4a yield was continually increased from 43% to 98% with the increase of temperature. Nevertheless, a decline of 3a yield occurred when the temperature was higher than 120℃. Clearly, 120℃ was the optimized reaction temperature.

|
Download:
|
| Fig. 1. Effect of (a) CO2 pressure and (b) reaction temperature on the product yields. Reaction conditions: 1a (76.1 mg, 1 mmol), 2a (84.1 mg, 1 mmol), DBU (76.0 mg, 0.5 mmol), DMF (2 mL), 10 h. (a) 120℃; (b) CO2 (3 MPa). The yield was determined by GC using biphenyl as the internal standard. 3a yield was based on 1a, and 4a yield was based on 2a. | |
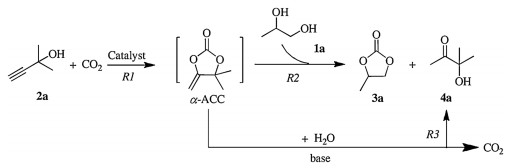
According to the reported works [31, 34], this three-component reaction proceeded with a sequential carboxylation and cyclization of propargylic alcohol and CO2 (R1, Scheme 2), and interesterification between intermediate α-alkylidene cyclic carbonate (α-ACC) and 1, 2-diol (R2) with the generation of cyclic carbonate and α-hydroxyl ketone. In addition, α-ACC was easily hydrolyzed to α-hydroxyl ketone in a basic condition (R3). Therefore, there was a competitive relationship between R2 and R3. As seen from Fig. 1b, increase temperature from 80℃ to 120℃ narrowed the gap in yield between 4a and 3a. Presumably, although the reaction rates of R2 and R3 were both accelerated with increasing temperature, R2 was signally promoted in the presence of the strong base DBU. In contrast, further increase in temperature led to a weaker catalytic activity, and the reaction rate of R3 was faster than that of R2. Accordingly, the yield of 4a was getting higher than that of 3a from 120 to 160℃. Additionally, an increased CO2 pressure might inhibit R3, leading to the decreased yield of 4a (Fig. 1a).

|
Download:
|
| Scheme 2. The procedure of the three-component cascade reaction. | |
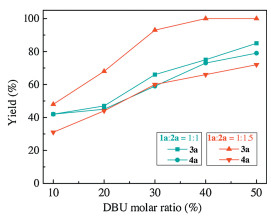
Fig. 2 showed the relationship between product yields and the amount of DBU. Based on the above conditions, increase the amount of DBU was found to be effective in facilitating the reaction process. However, both yields of 3a and 4a did not exceed 85%. However, further addition of 2a (molar ratio up to 1.5) greatly enhanced the catalytic efficiency, and 4a yield was up to 100% with 40 mol% DBU. These results indicated that increase the amount of propargylic alcohol in the catalytic system could promote R1 and further accelerate R2.

|
Download:
|
| Fig. 2. Dependence of the yield on amount of DBU. Reaction conditions: 1a (76.1 mg, 1 mmol), 2a (84.1 mg, 1 mmol)/(126.2 mg, 1.5 mmol), DMF (2 mL), CO2 (3 MPa), 120℃, 10 h. The yield was determined by GC using biphenyl as the internal standard. 3a yield was based on 1a, and 4a yield was based on 2a. | |
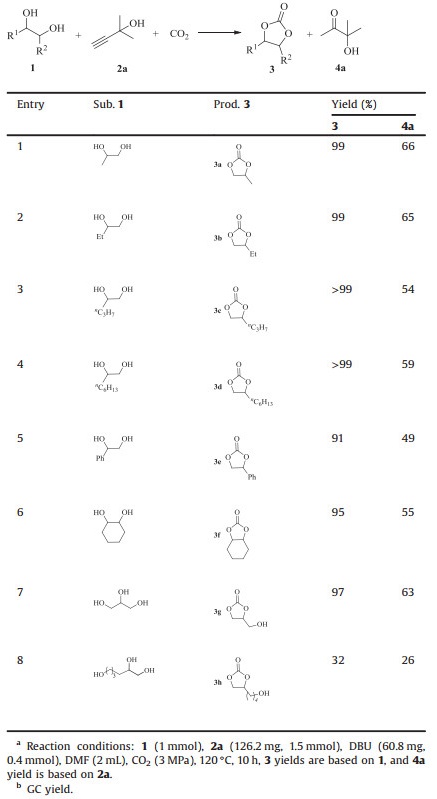
With the optimized reaction conditions in hand, the substrate scope was explored with 1.5 equiv. of propargylic alcohol at 120℃ under 3 MPa CO2 using 40 mol% of DBU as the catalyst in DMF for 10 h. As shown in Table 2, a series of vicinal diols were demonstrated to be effective under the identical conditions. The vicinal diols bearing different linear alkyl reacted with propargylic alcohol 2a and CO2, and gave cyclic carbonates 3a‒d and α-hydroxy ketone 4a in almost quantitative yields (entries 1–4). The arylsubstituted diol and 1, 2-cyclohexanediol showed a slight lower reactivity than that of alkyl-substituted diols, which indicated the influence of electronic and steric effect (entries 5 and 6). What is more, glycerol was also tested, and the target product, i.e., glycerol carbonate 3g was obtained in excellent yield (entry 7). 1, 2, 6-Hexanetriol was found to be effectively transformed into 3h, but its yield was lower than that of other carbonates (entry 8).
|
|
Table 2 Scope investigation for the reaction of vicinal diols 1, propargylic alcohol 2a, and CO2. a, b |
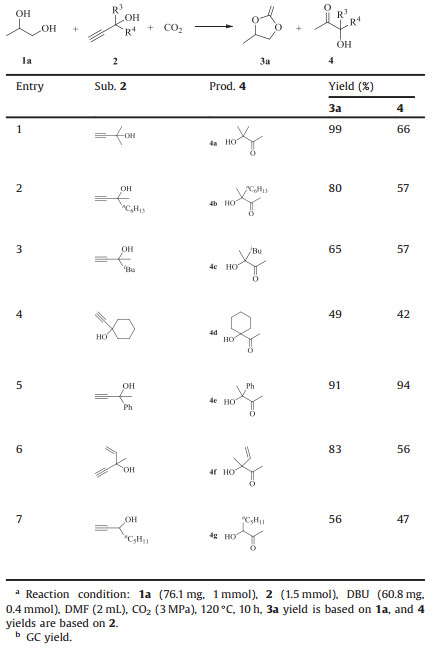
Next, we examined the effect of propargylic alcohols on the reaction (Table 3). As shown, all alkyl substituted propargylic alcohols could react with CO2 and 1a (entries 1–4), but a slightly lower yield was obtained when there was a large substituent group, such as isobutyl (entry 3) and cyclohexyl (entry 4). The reason might be due to the steric effect. Gratifyingly, the catalytic system worked well for the aryl-or vinyl-substituted propargylic alcohol (entries 5 and 6). In addition, it was found that the corresponding product 4g was also obtained in moderate yield when secondary alcohol was used as a substrate (entry 7).
|
|
Table 3 Scope investigation for the reaction of propylene glycol 1a, propargylic alcohol 2, and CO2. a, b |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
On the basis of previous reports [31, 32, 35-37, 45], a possible mechanism is proposed in Scheme 3. The first step is the carboxylation of activated propargylic alcohols with DBUCO2 adduct along with the formation of the intermediate A. An intramolecular ring-closing reaction of A will proceed and afford α-ACCs (This intermediate was demonstrated by control experiments in Scheme S1, Supporting information). Then, 1, 2-diols nucleophilic attack α-ACCs to form intermediate B, which subsequently transforms into intermediate C. Finally, cyclic carbonates and α-hydroxyl ketones are simultaneously obtained through intramolecular nucleophilic cyclization of intermediate C. Notably, hydrolysis of α-ACCs with trace water in a basic condition also generates α-hydroxyl ketones, which competes with main reaction.

|
Download:
|
| Scheme 3. Plausible mechanism. | |
{kind=link}
In summary, an efficient strategy for the synthesis of cyclic carbonates and α-hydroxy ketones through a three-component reaction of vicinal diols, propargylic alcohols, and CO2 in the presence of DBU catalysthasbeendeveloped.The resourceutilizationof CO2 through easily available vicinal diols and propargylic alcohols works efficiently. Through the activation of CO2 and substrates, the desired cyclic carbonates and α-hydroxy ketones are generated in good yield and selectivity. The mechanism revealed the main and side processes under the metal-free conditions.
AcknowledgmentsFinancial support from the National Natural Science Foundation of China (No. 21602232) and the Natural Science Foundation of Shanxi Province (No. 201701D221057) are gratefully acknowledged.
Appendix A. Supplementary dataSupplementarymaterial related to this article can befound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.06.030.
| [1] |
M. He, Y. Sun, B. Han, Angew. Chem. Int. Ed. 52 (2013) 9620-9633. DOI:10.1002/anie.201209384 |
| [2] |
M. Aresta, A. Dibenedetto, A. Angelini, Chem. Rev. 114 (2014) 1709-1742. DOI:10.1021/cr4002758 |
| [3] |
M. Cokoja, C. Bruckmeier, B. Rieger, W.A. Herrmann, F.E. Kühn, Angew. Chem. Int. Ed. 50 (2011) 8510-8537. DOI:10.1002/anie.201102010 |
| [4] |
Q. Liu, L. Wu, R. Jackstell, M. Beller, Nat. Commun. 6 (2015) 5933-5948. DOI:10.1038/ncomms6933 |
| [5] |
A. Otto, T. Grube, S. Schiebahn, D. Stolten, Energy Environ. Sci. 8 (2015) 3283-3297. DOI:10.1039/C5EE02591E |
| [6] |
B. Yu, L.N. He, ChemSusChem 8 (2015) 52-62. DOI:10.1002/cssc.201402837 |
| [7] |
T.E. Muller, W. Leitner, Beilstein J. Org. Chem. 11 (2015) 675-677. DOI:10.3762/bjoc.11.76 |
| [8] |
X.B. Lu, D.J. Darensbourg, Chem. Soc. Rev. 41 (2012) 1462-1484. DOI:10.1039/C1CS15142H |
| [9] |
K. Huang, C.L. Sun, Z.J. Shi, Chem. Soc. Rev. 40 (2011) 2435-2452. DOI:10.1039/c0cs00129e |
| [10] |
M. Cokoja, M.E. Wilhelm, M.H. Anthofer, W.A. Herrmann, F.E. Kuhn, ChemSusChem 8 (2015) 2436-2454. DOI:10.1002/cssc.201500161 |
| [11] |
G. Fiorani, W. Guo, A.W. Kleij, Green Chem. 17 (2015) 1375-1389. DOI:10.1039/C4GC01959H |
| [12] |
Y. Kayaki, M. Yamamoto, T. Ikariya, Angew. Chem. Int. Ed. 48 (2009) 4194-4197. DOI:10.1002/anie.200901399 |
| [13] |
A. Ueno, Y. Kayaki, T. Ikariya, Green Chem. 15 (2013) 425-430. DOI:10.1039/C2GC36414J |
| [14] |
K.I. Fujita, A. Fujii, J. Sato, S.Y. Onozawa, H. Yasuda, Tetrahedron Lett. 57 (2016) 1282-1284. DOI:10.1016/j.tetlet.2016.02.027 |
| [15] |
O. Jacquet, C. Das Neves Gomes, M. Ephritikhine, T. Cantat, J. Am. Chem. Soc. 134 (2012) 2934-2937. DOI:10.1021/ja211527q |
| [16] |
Y.B. Wang, Y.M. Wang, W.Z. Zhang, X.B. Lu, J. Am. Chem. Soc. 135 (2013) 11996-12003. DOI:10.1021/ja405114e |
| [17] |
Y.B. Wang, D.S. Sun, H. Zhou, W.Z. Zhang, X.B. Lu, Green Chem. 17 (2015) 4009-4015. DOI:10.1039/C5GC00948K |
| [18] |
C.M. Mömming, E. Otten, G. Kehr, et al., Angew. Chem. Int. Ed. 48 (2009) 6643-6646. DOI:10.1002/anie.200901636 |
| [19] |
T. Wang, D.W. Stephan, Chem. Eur. J. 20 (2014) 3036-3039. DOI:10.1002/chem.201304870 |
| [20] |
A.E. Ashley, A.L. Thompson, D. O'Hare, Angew. Chem. Int. Ed. 48 (2009) 9839-9843. DOI:10.1002/anie.200905466 |
| [21] |
N. von Wolff, G. Lefèvre, J.C. Berthet, P. Thuéry, T. Cantat, ACS Catal. 6 (2016) 4526-4535. DOI:10.1021/acscatal.6b00421 |
| [22] |
Z. Xin, C. Lescot, S.D. Friis, K. Daasbjerg, T. Skrydstrup, Angew. Chem. Int. Ed. 54 (2015) 6862-6866. DOI:10.1002/anie.201500233 |
| [23] |
X. Frogneux, E. Blondiaux, P. Thuéry, T. Cantat, ACS Catal. 5 (2015) 3983-3987. DOI:10.1021/acscatal.5b00734 |
| [24] |
E. Blondiaux, J. Pouessel, T. Cantat, Angew. Chem. Int. Ed. 53 (2014) 12186-12190. DOI:10.1002/anie.201407357 |
| [25] |
X.D. Lang, Y.C. Yu, Z.M. Li, L.N. He, J. CO2. Util. 15 (2016) 115-122. DOI:10.1016/j.jcou.2016.03.002 |
| [26] |
X. Gao, B. Yu, Z. Yang, et al., ACS Catal. 5 (2015) 6648-6652. DOI:10.1021/acscatal.5b01874 |
| [27] |
M. Liu, L. Liang, X. Li, X. Gao, J. Sun, Green Chem. 18 (2016) 2851-2863. DOI:10.1039/C5GC02605A |
| [28] |
Y.B. Wang, D.S. Sun, H. Zhou, W.Z. Zhang, X.B. Lu, Green Chem. 16 (2014) 2266-2272. DOI:10.1039/C3GC42346H |
| [29] |
H. Zhou, G.X. Wang, W.Z. Zhang, X.B. Lu, ACS Catal. 5 (2015) 6773-6779. DOI:10.1021/acscatal.5b01409 |
| [30] |
M.A. Courtemanche, M.A. Légaré, É. Rochette, F.G. Fontaine, Chem. Commun. (Camb.) 51 (2015) 6858-6861. DOI:10.1039/C5CC01282A |
| [31] |
Z.H. Zhou, Q.W. Song, L.N. He, ACS Omega 2 (2017) 337-345. DOI:10.1021/acsomega.6b00407 |
| [32] |
J.Y. Li, L.H. Han, Q.C. Xu, et al., ACS Sustainable Chem. Eng. 7 (2019) 3378-3388. DOI:10.1021/acssuschemeng.8b05579 |
| [33] |
X.D. Li, Y. Cao, R. Ma, L.N. He, J. CO2. Util 25 (2018) 338-345. DOI:10.1016/j.jcou.2018.01.022 |
| [34] |
Q.W. Song, Q.N. Zhao, J.Y. Li, K. Zhang, P. Liu, Synthesis 51 (2018) 739-746. |
| [35] |
J. Sun, W. Cheng, Z. Yang, et al., Green Chem. 16 (2014) 3071-3078. DOI:10.1039/c3gc41850b |
| [36] |
R. Nicholls, S. Kaufhold, B.N. Nguyen, Catal. Sci. Technol. 4 (2014) 3458-3462. DOI:10.1039/C4CY00480A |
| [37] |
Z.Z. Yang, Y.N. Zhao, L.N. He, J. Gao, Z.S. Yin, Green Chem. 14 (2012) 519-527. DOI:10.1039/c2gc16039k |
| [38] |
K. Chen, G. Shi, R. Dao, et al., Chem. Commun. (Camb.) 52 (2016) 7830-7833. DOI:10.1039/C6CC02853E |
| [39] |
N.D. Ca, B. Gabriele, G. Ruffolo, et al., Adv. Synth. Catal. 353 (2011) 133-146. DOI:10.1002/adsc.201000607 |
| [40] |
M. Baidya, H. Mayr, Chem. Commun. (Camb.) (2008) 1792-1794. |
| [41] |
M. Hulla, S.M.A. Chamam, G. Laurenczy, S. Das, P.J. Dyson, Angew. Chem. 129 (2017) 10695-10699. DOI:10.1002/ange.201705438 |
| [42] |
T. Mizuno, Y. Ishino, Tetrahedron 58 (2002) 3155-3158. DOI:10.1016/S0040-4020(02)00279-X |
| [43] |
P.G. Jessop, B. Subramaniam, Chem. Rev. 107 (2007) 2666-2694. DOI:10.1021/cr040199o |
| [44] |
C. Roosen, M. Ansorge-Schumacher, T. Mang, W. Leitner, L. Greiner, Green Chem. 9 (2007) 455-458. DOI:10.1039/b613345b |
| [45] |
E.R. Pérez, R.H.A. Santos, M.T.P. Gambardella, et al., J. Org. Chem. 69 (2004) 8005-8011. DOI:10.1021/jo049243q |