 2020, Vol. 31
2020, Vol. 31 


b Shanghai Engineering Research Center of Molecular Therapeutics and New Drug Development, School of Chemistry and Molecular Engineering, East China Normal University, Shanghai 200062, China;
c State Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai 200032, China
The utilization of carbon dioxide (CO2) as a renewable, abundant and nontoxic C1 feedstock for chemical synthesis has received considerable attentions [1]. As it not only provides an attractive complement to carbon capture and storage, but also presents an important way to realize the green carbon science [2].Over the past decades, a series of reactions have been developed for CO2 chemical transformation [3], among which, the carboxylative cyclization of propargylic alcohols and CO2 is very attractive [4], as the thus obtained α-alkylidene cyclic carbonates have wide applications in both organic synthesis and industrial chemistry [5].However, duo to the well-known challenge associate with enantioselective transformation of CO2, such as thermodynamic stability and kinetic inertness, the catalytic asymmetric carboxylative cyclization of propargylic alcohols and CO2 is largely undeveloped [6]. The only two successful reports were realized by Yoshida & Ihara and Yamada respectively [7]. As early as 2003, Yoshida & Ihara developed a palladium-catalyzed cascade reaction of 4-methoxycarbonyloxy-2-butyn-1-ols with phenols and CO2, which involved a CO2 elimination-fixation process (Scheme 1A) [7a]. In 2010, Yamada and co-workers reported an enantioselective carboxylative cyclization of propargylic alcohols with CO2 via a chiral silver catalyzed desymmetrization process (Scheme 1B) [7b]. Distinct from those researches, we envisioned that enantioselective kinetic resolution would be another promising strategy for the development of enantioselective carboxylative cyclization of propargylic alcohols and CO2 (Scheme 1C) [8]. The attractive features of this approach include the easy accessibility of various types of propargylic alcohols and, more importantly, the chiral cyclic carbonates and optically active propargylic alcohols could be obtained simultaneously under mild conditions.

|
Download:
|
| Scheme 1. Asymmetric carboxylative cyclization of propargylic alcohol and CO2. | |
The optically active propargylic alcohols are highly important as versatile building blocks for the construction of biologically active natural products as well as pharmaceuticals [9]. Enormous efforts have been devoted to the development of highly efficient catalytic asymmetric methods for propargylic alcohol synthesis [10].However, most of these reports are aim to the chiral secondary propargylic alcohols, the access to optically active tertiary propargylic alcohols is much more challenging [11]. In this context, the development of new synthetic methods to optically active tertiary propargylic alcohols is highly desirable.
In light of these facts, together with our continue efforts in the enantioselective construction of chiral tertiary alcohols [12] and the chemical fixation of CO2 for value-added products [13], herein, we wish to report our preliminary research in developing a catalytic asymmetric carboxylative cyclization of propargylic alcohols and CO2 for the synthesis of optical active tertiary propargylic alcohols and chiral cyclic carbonates simultaneously, based on a kinetic resolution strategy.
The 1-indanone derived propargylic alcohol 1a was utilized for the investigation of the reaction, as it could give the novel spirocyclic carbonates and the chiral cyclic propargylic alcohols simultaneously, both compounds have a range of applications. We initiated our research by studying the kinetic resolution under 1 atm pressure of CO2 in the presence of 10 mol% of AgOAc with various chiral ligands (for detail, see Supporting information). To our delight, the 1-(2-pyridinyl)ethanone derived chiral Schiff base L1, which had been used by Yamada in the desymmetric carboxylative cyclization [7b], could promoted the reaction smoothly in dichloromethane (DCM) under room temperature, to give the desired chiral carbonate 2a in 49% yield and 30% ee, with the recovery of optical active 1a in 46% yield and 43% ee (entry 1, Table 1). Other chiral ketimine ligand, such as L2, L3 and L4 bearing a 4-sulfuryl, 5-trifluoromethyl or 6-bromo group in pyridyl ring, gave slightly lower yield and enantioslectivity respectively (entries 2-4). Then, we turned attention to the 2-pyridinecarboxyaldehyde derived Schiff base ligand, and found that the substituents on the pyridyl ring had great influence on the reaction. Although the simple chiral aldimine ligand L5 gave very poor results, the enantioselectivity increased gradually when the 6-substituted chiral ligands L5-L9 was employed (entries 5-9). Finally, chiral ligand L8 with an electron-withdrawing 6-chloro-substituted group was demonstrated to be the best one, affording the chiral 2a in 53% yield and 36% ee, with the recovery of chiral 1a in 46% yield and 60% ee (entry 8). The performance of other types of chiral ligand such as Bn-PYBOX was also studied, but only 3% ee for both 1a and 2a were obtained (entry 10).
|
|
Table 1 Ligand effects. |

To further improve the reaction outcomes, various silver salts and solvents were briefly investigated by using Schiff base L8 as chiral ligand. As shown in Table 2, the silver salts had dramatic effects on the reaction. Apart from AgOAc and AgOBz, other ones failed to catalyze the reaction. While AgOBz gave slightly diminished enantioselectivity than AgOAc (entries 1-2, Table 2).The solvent effects were also investigated, but no better results could be obtained (entries 3-5). Therefore, the substrate scope was then examined by using L8 /AgOAc complex as catalyst, DCM as solvent, under 1 atmosphere of CO2 pressure.
|
|
Table 2 Effects of silver salts and solvents. |
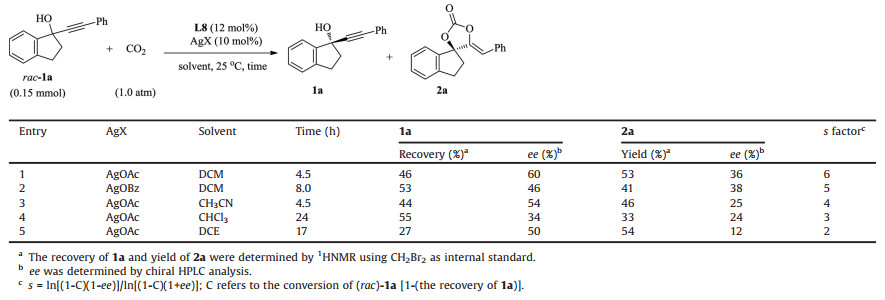
As shown in Table 3, most of the 1-indanone derived propargylic alcohols bearing different substituents on the alkynyl moiety or phenyl ring were viable substrates for this kinetic resolution, with moderate enantioselectivity achieved. Initially, propargylic alcohols with different substituents (R2) on the alkynyl moiety were investigated. As expected, with the R2 group varying from phenyl to para-halogenated phenyl groups, the reaction rates increased to a certain degree, and the carbonates 2b-2d could be obtained smoothly, with the recovery of the chiral propargylic alcohols 1b-1d in 40%-50% yields and 50%-59% ee values (entries 2-4). For the reaction of para-methylphenyl substituted propargylic alcohols, affected by the electron-donating effect, a slightly higher 0.5 MPa CO2 pressure was needed (entry 5). By verifying the substituent in the phenyl ring from para-position to meta-position, the reaction could also proceed well to give the corresponding chiral carbonates 2f-2h in 38%-49% yield, with the recovery of chiral alcohols 1f-1h in 43%-59% yield and 37%-54% ee value (entries 6-8). When ortho-fluoro-or ortho-chloro-phenyl substituted propargylic alcohol 2i or 2 j was applied to the reaction, a slightly lower reaction rate was observed, probably duo to the steric hindrance effect (entries 9-10). Then, the reaction of propargylic alcohols 1k and 1l bearing 4-bromo and 5-fluoro substituent at the phenyl ring were studied, which could deliver the corresponding chiral carbonate 2k and 2l in 58% and 49% yield, along with the recovery of enantioenriched propargylic alcohol 1k and 1l in 42% and 51% yield with 45% and 47% ee respectively (entries 11 and 12). Further attempts to use the alkyl substituted or terminal alkyne derived propargylic alcohols were nevertheless unsuccessful, and no desired products could be detected. The absolute configuration of chiral alcohol 1a was determined to be S by comparing its optical rotation value with the one synthesized by the asymmetric nucleophilic addition reaction [14]. Based on this, the absolute configuration of other chiral propargylic alcohols and carbonates were tentatively deduced by analogy (for detail, see Supporting information).
|
|
Table 3 The substrate scope. |
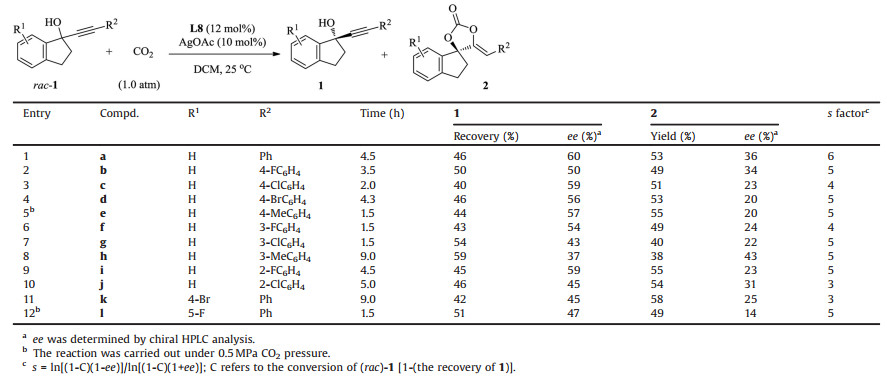
It should be point out that in most cases of the reaction, there is a notable difference between the ee value of recovered alcohols and chiral carbonates, even though the conversions are about 50% and the transformations are very clean. Theoretically, the ee value of both chiral compounds should be very similar to each other. To figure out the reason, the reaction of racemic-1f was monitored over time. As shown in Table 4, with the reaction went on, the conversion and the ee value of 1f increased gradually, but the ee value of 2f went up to a certain value, then dropped down. These results indicated that the chiral carbonate might be racemized during the reaction cause, and the best results could be obtained at around 50% conversion.
|
|
Table 4 The monitor of the reaction course. |
{kind=link}
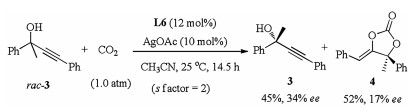
Finally, the reaction of the non-cyclic propargylic alcohol 3 with CO2 was studied (Scheme 2), by using the L6/AgOAc as chiral catalyst and CH3CN as solvent, the chiral carbonate 4 was obtained in 52% yield, with the recovery of chiral propargylic alcohols 3 in 45% yield and 34% ee value.

|
Download:
|
| Scheme 2. The kinetic resolution of non-cyclic propargylic alcohol. | |
{kind=link}
In conclusion, we have developed a novel catalytic asymmetric carboxylative cyclization of propargylic alcohols with CO2 based on a kinetic resolution process. Under mild reaction conditions, both optically active propargylic alcohols and chiral cyclic carbonates could be obtained simultaneously with considerable yield and moderate enantioselectivity by simple operation. Further investigation including the exploitation of new catalytic systems to improve the catalytic efficiency along with the development of other catalytic asymmetric reactions using CO2 as C1 synthon are now in progress in our laboratory.
AcknowledgmentsThe financial support from 973 Program (No. 2015CB856600), the National Natural Science Foundation of China (Nos. 21871090, 21573073) and the Fundamental Research Funds for the Central Universities is highly appreciated.
Appendix A. Supplementary dataSupplementarymaterial related to this article canbefound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.05.060.
| [1] |
(a) T. Sakakura, J.C. Choi, H. Yasuda, Chem. Rev. 107 (2007) 2365-2387; (b) D.M. D'Alessandro, B. Smit, J.R. Long, Angew. Chem. Int. Ed. 49 (2010) 6058-6082; (c) M. Cokoja, C. Bruckmeier, B. Rieger, W.A. Herrmann, F.E. Kühn, Angew. Chem. Int. Ed. 50 (2011) 8510-8537; (d) E.V. Kondratenko, G. Mul, J. Baltrusaitis, G.O. Larraźabalc, J. Pérez-Ramírez, Energy Environ. Sci. 6 (2013) 3112-3135; (e) M. Aresta, A. Dibenedetto, A. Angelini, Chem. Rev. 114 (2014) 1709-1742; (f) X.B. Lu, Carbon Dioxide and Organometallics, Springer, Switzerland, (2016); (g) A.W. Kleij, M. North, A. Urakawa, ChemSusChem 10 (2017) 1036-1038; (h) S.S. Yan, Q. Fu, L.L. Liao, et al., Coord. Chem. Rev. 374 (2018) 439-463; (i) S. Wang, C. Xi, Chem. Soc. Rev. 48 (2019) 382-404. |
| [2] |
M. He, Y. Sun, B. Han, Angew. Chem. Int. Ed. 52 (2013) 9620-9633. DOI:10.1002/anie.201209384 |
| [3] |
(a) Q. Liu, L. Wu, R. Jackstell, M. Beller, Nat. Commun. 6 (2015) 5933-5947; (b) C. Maeda, Y. Miyazaki, T. Ema, Catal. Sci. Technol. 4 (2014) 1482-1497; (c) G. Fiorani, W. Guo, A.W. Kleij, Green Chem. 17 (2015) 1375-1389; (d) M. Börjesson, T. Moragas, D. Gallego, R. Martin, ACS Catal. 6 (2016) 6739-6749; (e) S.N. Riduan, Y. Zhang, Dalton Trans. 39 (2010) 3347-3357; (f) W. Zhang, C. Guo, X. Lü, Chin. J. Catal. 37 (2016) 215-217; (g) Z. Zhang, T. Ju, J.H. Ye, D.G. Yu, Synlett 28 (2017) 741-750; (h) W. Zhang, N. Zhang, C. Guo, X. Lü, Chin. J. Org. Chem. 37 (2017) 1309-1321; (i) Y.Y. Gui, W.J. Zhou, J.H. Ye, D.G. Yu, ChemSusChem 10 (2017) 1337-1340; (j) Y. Cao, X. He, N. Wang, H.R. Li, L.N. He, Chin. J. Chem. 36 (2018) 644-659; (k) Z. Zhang, J.H. Ye, D.S. Wu, Y.Q. Zhou, D.G. Yu, Chem. -Asian J. 13 (2018) 2292-2306. |
| [4] |
(a) J.W. Comerford, I.D.V. Ingram, M. North, X. Wu, Green Chem. 17 (2015) 1966-1987; (b) Q.W. Song, L.N. He, Adv. Synth. Catal. 358 (2016) 1251-1258; (c) B. Zou, Chin. J. Chem. 35 (2017) 541-550; (d) Y. Wu, Y. Zhao, R. Li, et al., ACS Catal. 7 (2017) 6251-6255; (e) H. Zhou, G.X. Wang, X.B. Lu, Asian J. Org. Chem. 6 (2017) 1264-1269; (f) S. Sun, B. Wang, N. Gu, J.T. Yu, J. Cheng, Org. Lett. 19 (2017) 1088-1091; (g) G. Shen, W.J. Zhou, X.B. Zhang, et al., Chem. Commun. 54 (2018) 5610-5613. |
| [5] |
H. Zhang, H.B. Liu, J.M. Yue, Chem. Rev. 114 (2014) 883-898. DOI:10.1021/cr300430e |
| [6] |
(a) J. Vaitla, Y. Guttormsen, J.K. Mannisto, et al., ACS Catal. 7 (2017) 7231-7244; (b) N. Kielland, C.J. Whiteoak, A.W. Kleija, Adv. Synth. Catal. 355 (2013) 2115-2138; (c) X.B. Lu, B. Liang, Y.J. Zhang, et al., J. Am. Chem. Soc. 126 (2004) 3732-3733; (d) M. Takimoto, Y. Nakamura, K. Kimura, M. Mori, J. Am. Chem. Soc. 126 (2004) 5956-5957; (e) M. Zhang, X. Zhao, S. Zheng, Chem. Commun. 50 (2014) 4455-4458; (f) B.A. Vara, T.J. Struble, W. Wang, M.C. Dobish, J.N. Johnston, J. Am. Chem. Soc. 137 (2015) 7302-7305; (g) Y.Y. Gui, N. Hu, X.W. Chen, et al., J. Am. Chem. Soc. 139 (2017) 17011-17014. |
| [7] |
(a) M. Yoshida, M. Fujita, T. Ishii, M. Ihara, J. Am. Chem. Soc. 125 (2003) 4874-4881; (b) S. Yoshida, K. Fukui, S. Kikuchi, T. Yamada, J. Am. Chem. Soc. 132 (2010) 4072-4073. |
| [8] |
(a) J.M. Keith, J.F. Larrow, E.N. Jacobsen, Adv. Synth. Catal. 343 (2001) 5-26; (b) E. Vedejs, M. Jure, Angew. Chem. Int. Ed. 44 (2005) 3974-4001; (c) H. Pellissier, Adv. Synth. Catal. 353 (2011) 1613-1666; (d) C.E. Müller, P.R. Schreiner, Angew. Chem. Int. Ed. 50 (2011) 6012-6042; (e) R. Gurubrahamam, Y.S. Cheng, W.Y. Huang, K. Chen, ChemCatChem 8 (2016) 86-96; (f) O. Verho, J.E. Backvall, J. Am. Chem. Soc. 137 (2015) 3996-4009; (g) G. Ma, M.P. Sibi, Chem. -Eur. J. 21 (2015) 11644-11657. |
| [9] |
(a) Y. Zhu, L. Sun, P. Lu, Y. Wang, ACS Catal. 4 (2014) 1911-1925; (b) Q. Wang, L. Pu, Synlett 24 (2013) 1340-1363; (c) P.J. Stang, F. Diederich, Modern Acetylene Chemistry, Wiley-VCH: Weinheim, 2008. |
| [10] |
(a) Q.W. He, S.M. Ma, Chinese J. Org. Chem. 22 (2002) 375-387; (b) L. Pu, Tetrahedron 59 (2003) 9873-9886; (c) G. Lu, Y.M. Li, X.S. Li, A.S.C. Chan, Coordin. Chem. Rev. 249 (2005) 1736-1744; (d) B.M. Trost, A.H. Weiss, Adv. Synth. Catal. 351 (2009) 963-983; (e) B.M. Trost, M.J. Bartlett, A.H. Weiss, A.J.V. Wangelin, V.S. Chan, Chem. -Eur. J. 18 (2012) 16498-16509; (f) V. Bisai, V.K. Singh, Tetrahedron Lett. 57 (2016) 4771-4784; (g) H. Noda, N. Kumagai, M. Shibasaki, Asian J. Org. Chem. 7 (2018) 599-612. |
| [11] |
(a) B. Jiang, Z. Chen, X. Tang, Org. Lett. 4 (2002) 3451-3453; (b) P.G. Cozzi, Angew. Chem. Int. Ed. 42 (2003) 2895-2898; (c) G. Lu, X. Li, X. Jia, W.L. Chan, A.S.C. Chan, Angew. Chem. Int. Ed. 42 (2003) 5057-5058; (d) P.G. Cozzi, S. Alesi, Chem. Commun. (2004) 2448-2449; (e) C. Chen, L. Hong, Z.Q. Xu, L. Liu, R. Wang, Org. Lett. 8 (2006) 2277-2280; (f) K. Tanaka, K. Kukita, T. Ichibakase, S. Kotanib, M. Nakajima, Chem. Commun. 47 (2011) 5614-5616; (g) T. Ohshima, T. Kawabata, Y. Takeuchi, et al., Angew. Chem. Int. Ed. 50 (2011) 6296-6300; (h) T. Wang, J.L. Niu, S.L. Liu, et al., Adv. Synth. Catal. 355 (2013) 927-937; (i) P. Smirnov, J. Mathew, A. Nijs, et al., Angew. Chem. Int. Ed. 52 (2013) 13717-13721; (j) Y. Zheng, Y. Tan, K. Harms, et al., J. Am. Chem. Soc. 139 (2017) 4322-4325; (k) B.P. Zavesky, J.S. Johnson, Angew. Chem. Int. Ed. 56 (2017) 8805-8808; (l) M.C. Schwarzer, A. Fujioka, T. Ishii, et al., Chem. Sci. 9 (2018) 3484-3493; (m) D.K. Friel, M.L. Snapper, A.H. Hoveyda, J. Am. Chem. Soc.130 (2008) 9942-9951; (n) H. Kawai, K. Tachi, E. Tokunaga, M. Shiro, N. Shibata, Org. Lett. 12 (2010) 5104-5107; (o) H. Noda, F. Amemiya, K. Weidner, N. Kumagai, M. Shibasaki, Chem. Sci. 8 (2017) 3260-3269; (p) W. Zhang, S. Ma, Chem. Commun. 54 (2018) 6064-6067. |
| [12] |
(a) Z.Y. Cao, F. Zhou, J. Zhou, Acc. Chem. Res. 51 (2018) 1443-1454; (b) Y.L. Liu, X.P. Yin, J. Zhou, Chin. J. Chem. 36 (2018) 321-328; (c) X.P. Zeng, J. Zhou, J. Am. Chem. Soc. 138 (2016) 8730-8733; (d) Z.Y. Cao, J.S. Jiang, J. Zhou, Org. Biomol. Chem. 14 (2016) 5500-5504; (e) X.P. Zeng, Z.Y. Cao, X. Wang, et al., J. Am. Chem. Soc. 138 (2016) 416-425; (f) Y.L. Liu, F.M. Liao, Y.F. Niu, X.L. Zhao, J. Zhou, Org. Chem. Front.1 (2014) 742-747; (g) Y.L. Liu, J. Zhou, Acta Chim. Sinica 70 (2012) 1451-1456; (h) Y.L. Liu, J. Zhou, Chem. Commun. 48 (2012) 1919-1921; (i) Z.Y. Cao, Y. Zhang, C.B. Ji, J. Zhou, Org. Lett. 13 (2011) 6398-6401; (j) Y.L. Liu, B.L. Wang, J.J. Cao, et al., J. Am. Chem. Soc.132 (2010) 15176-15178; (k) J.J. Cao, F. Zhou, J. Zhou, Angew. Chem. Int. Ed. 49 (2010) 4976-4980. |
| [13] |
(a) X.T. Gao, C.C. Gan, S.Y. Liu, et al., ACS Catal. 7 (2017) 8588-8593; (b) F. Zhou, S.L. Xie, X.T. Gao, et al., Green Chem. 19 (2017) 3908-3915. |
| [14] |
(a) S. Kotani, K. Kukita, K. Tanaka, et al., J. Org. Chem. 79 (2014) 4817-4825; (b) B. Bieszczad, D.G. Gilheany, Angew. Chem. Int. Ed. 56 (2017) 4272-4276. |