 2020, Vol. 31
2020, Vol. 31 


b CCNU-uOttawa Joint Research Centre, Key Laboratory of Pesticides & Chemical Biology Ministry of Education, College of Chemistry, Central China Normal University, Wuhan 430079, China
Chiral indolines are an important class of nitrogen heterocycles because of their prevalence in many alkaloid natural products, pharmaceuticals, and biologically active compounds [1]. Of them, enantiomerically pure 2-substituted indolines have been utilized as chiral auxiliaries in asymmetric synthesis [2], therapeutic reagents [3] as well as a common motif of biologically significant compounds such as oleraceins A–D [4a], neuronal cell-protecting substances benzastatins E–G [4b], the antihypertensive drug Pentopril [4c], anticancer agent [4d], the antitumor agent [4e], and an oral antihypertensive/diuretic Indapamide [4f] (Fig. 1).

|
Download:
|
| Fig. 1. Representative examples of biologically active 2-substituted indoline derivatives. | |
{kind=link}
Given their extensive applications, chiral 2-substituted indolines have drawn the increasing interest of synthetic chemists [5]. Not surprisingly, in recent years, a number of efficient methods have been developed for the synthesis of these chiral compounds, including kinetic resolution, indole functionalization, asymmetric synthesis from chiral reagents, and asymmetric catalysis. This review concentrates on the typical strategies developed since 2012.
2. Kinetic resolutionKinetic resolution of racemates is an effective method for the preparation of enantiomerically pure compounds, which is most widely used in the industrial sector [6]. In this regard, many enzymatic and non-enzymatic protocols have been well developed for the enantioselective synthesis of 2-substituted indolines. Thus, in this section, some representative achievements in this context will be discussed.
2.1. Enzymatic kinetic resolutionStrategies employing enzymes as catalysts for kinetic resolution of enantiomers have been proved to be one of the most elegant and sustainable protocols in synthetic organic chemistry [7]. GotorFernández et al. developed an efficient chemoenzymatic process to afford 2, 3-disubstituted indoline diastereoisomers [8]. The combination of Candida antarctica lipase type A (CAL-A) and allyl-3-methoxyphenyl carbonate 2 in TBME (tert-butyl methyl ether) as a solvent at 30 ℃ and 250 rpm for the necessary time allowed the isolation of the carbamate 3 and indoline (2S, 3S)-1 with excellent enantioselectivity (Scheme 1).

|
Download:
|
| Scheme 1. Enzymatic kinetic resolution of (±)-cis-1 with CAL-A and carbonate 2. | |
{kind=link}
2.2. Non-enzymatic kinetic resolution
In addition to the kinetic resolutions based on the use of enzymes, transition-metal and organocatalyzed kinetic resolutions also gain popularity recently due to the progress made in the discovery of novel chiral catalysts. So far, many approaches based on the catalytic non-enzymatic kinetic resolution have been developed with high enantioselectivity [9].
In 2013, Akiyama's group disclosed an oxidative kinetic resolution of 2-substituted indoline derivatives 1 [10a]. This transformation was achieved by asymmetric transfer hydrogenation to aromatic imines 4 by means of chiral phosphoric acid 5a. A series of alkyl-or aryl-substituted indolines (2S, 3S)-1 were obtained in good yield with excellent enantioselectivity (Scheme 2a).

|
Download:
|
| Scheme 2. Oxidative kinetic resolutions catalyzed by chiral phosphoric acid for the synthesis of optically active indolines. | |
{kind=link}
Subsequently, the same group reported another oxidative kinetic resolution of 2-substituted indoline derivatives 8 based on a self-redox reaction with a chiral phosphoric acid catalyst 5b. This method featured an asymmetric intermolecular hydride transfer from indoline (S)-8 to iminium intermediate 10 generated from the condensation reaction of indoline (S)-8 with salicylaldehyde 9. The oxidative kinetic resolution afforded another enantiomers (R)-8 in high yield and excellent enantioselectivity (Scheme 2b) [10b].
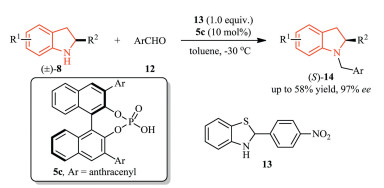
Moreover, Zhao et al. developed a protocol for obtaining the N-benzyl indolines (S)-14 via oxidative kinetic resolution of in situ formed aldiminium intermediate in good yield with moderate to excellent enantioselectivity [11]. Different parameters such as hydride donor, catalyst, molecular sieves, and temperature were screened in order to establish the optimal reaction conditions (Scheme 3).

|
Download:
|
| Scheme 3. Kinetic resolutions of indolines through reductive amination of aldehyde by indolines by chiral phosphoric acid. | |
{kind=link}
In 2017, Spivey and co-workers described a catalytic kinetic resolution of 2-substituted indoline derivatives by N-sulfonylation using an atropisomeric 2-aryl-4-DMAP-N-oxide catalyst (-)-16, affording the optically active 2-substituted indolines (S)-17 [12].The use of catalyst 16 in combination with 2-isopropyl-4-nitrophenylsulfonyl chloride 15 was crucial to achieve high enantioselectivity and facile deprotection of the sulfonamide products (S)- 17 could be easily realized with thioglycolic acid. A qualitative model 18 was established to rationalize the substrate scope for the stereodiscrimination (Scheme 4). The kinetic resolution of racemic indolines was extensively explored, mainly through acylation on the nitrogen atom [6].

|
Download:
|
| Scheme 4. N-Sulfonylative kinetic resolutions of indolines using an atropisomeric 4-DMAP-N-oxide organocatalyst. DIPEA: N, N-diisopropylethylamine. | |
{kind=link}
There are also some other impressive works in the field of kinetic resolution for the synthesis of chiral indolines. For instance, in 2013, Cai and co-workers found that the kinetic resolution of racemic 2-amino-3-(2-iodoaryl)propanoates 19 could be successfully achieved via Cu(I)/BINOL system-catalyzed intramolecular N-arylation [13] (Scheme 5). The reaction afforded chiral indolines (S)-21 bearing chiral carbon centers at C2 position, and the starting materials (R)-19 could be recovered with excellent enantioselectivity.

|
Download:
|
| Scheme 5. Kinetic resolution of racemic 2-amino-3-(2-iodoaryl)propanoates by intramolecular N-arylation via Cu(I)/BINOL system. | |
{kind=link}
In 2016, the Fan group found that a kinetic resolution of 3-H-indoles 22a, 22b bearing different substituents at C3 position could be realized by asymmetric hydrogenation [14]. Under the optimized reaction conditions, two hydrogenated products (2R, 3R)-24a (83% ee), (2R, 3R)-24b (64% ee) as well as two recovered chiral substrates (S)-22a and (S)-22b were obtained with excellent enantioselectivity (97% ee and 93% ee) (Scheme 6).

|
Download:
|
| Scheme 6. Kinetic resolution of racemic 3, 3-disubstituted 3H-indoles by asymmetric hydrogenation. HFIP: hexafluoroisopropanol. | |
{kind=link}
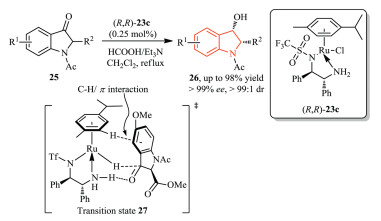
Zhang, Wang, and co-workers disclosed a Ru(Ⅱ)-catalyzed dynamic kinetic resolution-asymmetric transfer hydrogenation of racemic 2-substituted-N-acetyl-3-oxoindolines 25 to cis-2-substituted-N-acetyl-3-hydroxyindolines 26 (Scheme 7) [15]. This method provided 2-substituted N-acetyl-3-hydroxyindoline derivatives 26 as only cis diastereomers in high yield (up to 98% yield) with excellent diastereoselectivity (> 99:1 dr) and enantioselectivity (up to > 99% ee). This result might arise from the proposed transition state 27, wherein the orientation of the Ru-H catalyst 23c and the phenyl group of the substrate played an important role.

|
Download:
|
| Scheme 7. Ru(Ⅱ)-catalyzed dynamic kinetic resolution-asymmetric transfer hydrogenation of racemic 2-substituted-N-acetyl-3-oxoindolines. | |
{kind=link}
3. Indole functionalization 3.1. Asymmetric hydrogenation of indoles
Chiral palladium complex have been extensively applied in asymmetric hydrogenation of a large number of substrates, including heteroaromatics [16]. In 2012, Zhou and co-workers accomplished an efficient asymmetric hydrogenation reaction of 2-substituted 3-(toluenesulfonamidoalkyl)indoles 28 using a chiral Pd catalyst in the presence of a Brønsted acid TsOH (Scheme 8a) [17]. In this reaction, the key vinylogous iminium intermediate 31 was initially generated from indole substrate 28 in the presence of Brønsted acid, which then underwent an asymmetric hydrogenation reaction to afford the chiral 2, 3-disubstituted indolines 30 with up to 97% ee. Moreover, the one-pot tandem reaction (Friedel-Crafts/asymmetric hydrogenation) of 2-substituted indoles 32 also proceeded smoothly without erosion of the enantioselectivity (up to 95% ee) (Scheme 8b).

|
Download:
|
| Scheme 8. Pd-catalyzed asymmetric hydrogenation of 3-(toluenesulfonamidoalkyl)indoles and its tandem process. | |
{kind=link}
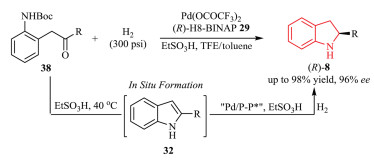
Taking advantage of this substrate activation strategy, Fan and Zhou et al. subsequently developed an efficient Pd-catalyzed asymmetric hydrogenation of an array of diversely substituted indoles 32 and 7 including 2-substituted and 2, 3-disubstituted indoles (Schemes 9a and b) [18]. This methodology was also applied in the synthesis of biologically active indoline skeleton, a neuroleptic agent 34. The mechanism of this process was extensively investigated by the control experiments and DFT (density functional theory) calculations. The isotope-labeling experiments and ESI-HRMS suggested that the involvement of the iminium intermediate should be formed in situ via the protonation of the C2 = C3 bond of simple unprotected indoles with Brønsted acid; this step was crucial for full conversion and high enantioselectivity in this reaction. This key intermediate was then hydrogenated by the chiral Pd catalyst through an outer-sphere mechanism involving stepwise proton and hydride transfers from active Pd-H species. The reaction took place well in highly polar solvents such as TFE (trifluoroethanol) due to its ability to stabilize the ionic intermediate 35 (Scheme 9c). The authors reported an asymmetric hydrogenation of unprotected indoles 7 by the same strategy with ligand 36 and EtSO3H as a Brønsted acid activator, although the results obtained, in terms of enantioselectivity, were not as good (up to 87% ee; Scheme 9d) as that with p-TsOH (up to 98% ee; Scheme 9b) [19].

|
Download:
|
| Scheme 9. Pd-catalyzed asymmetric hydrogenation of indoles. | |
{kind=link}
Shortly thereafter, Liu, Zhang and co-workers also reported the Pd-catalyzed enantioselective hydrogenation of differently substituted indoles 7 using an axially chiral diphosphine (S)-C10-BridgePhos ligand 37 with a large bite angle. With D-CSA (D-camphorsulfonic acid) as activator, chiral indoline products (2S, 3S)-1 were obtained in quantitative conversion and excellent enantioselectivity (up to 98% ee, Scheme 9e). The authors proposed that the substituent at 2-position played an important role in the hydrogenation process [20].
In 2018, Zhou's group accomplished a more concise and enantioselective route for the chiral indoline synthesis through a one-pot procedure, which involved a sequential intramolecular condensation, deprotection and palladium-catalyzed asymmetric hydrogenation [21]. Again, it was found that the Brønsted acid played a significant role in this process (Scheme 10). Notably, this methodology could be scaled up without obvious loss of reactivity and enantioselectivity.

|
Download:
|
| Scheme 10. Asymmetric hydrogenation of in situ generated indoles. | |
{kind=link}
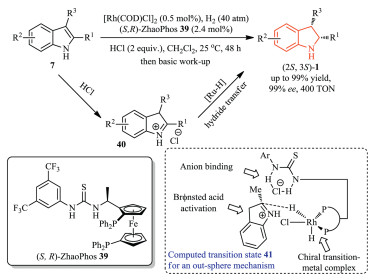
In 2018, Chung, Zhang and co-workers reported a Brønsted acid-promoted Rh-catalyzed asymmetric hydrogenation of N-unprotected indoles 7 (Scheme 11). Fluorinated alcohol was avoided in this reaction, which made this methodology more economic to chiral indolines with high enantioselectivity (up to 99% ee, up to 400 TON). Mechanistically, the reaction proceeded via the substrate activation with Brønsted acid, thiourea anion binding, and rhodium/ZhaoPhos complex. In addition, DFT calculations revealed an outer-sphere mechanism in this transformation [22].

|
Download:
|
| Scheme 11. Asymmetric hydrogenation of indoles with simple Brønsted acid: a cocatalysis of transition metal and anion binding. | |
{kind=link}
Despite the above-mentioned powerful methods, several challenges still remained: the use of stoichiometric amounts of Brønsted acid as the activator and the relatively high catalyst loading. In order to tackle these challenges, Vidal-Ferran and coworkers employed enantiomerically pure iridium complexe derived from their P-OP ligand 42 (relatively low loading: 1 mol%) and (easily recyclable and reusable) Brønsted acids for the efficient asymmetric hydrogenation of unprotected indoles, which allowed the formation of optically pure indolines with up to 78% yield and 91% ee (Scheme 12) [23]. Remarkably, the DOWEXTM 50WX8 resin could be recovered, recycled, and reused for twice, showing comparable catalytic activity.

|
Download:
|
| Scheme 12. Ir-catalyzed asymmetric hydrogenation of indoles with reusable Brønsted acid. THF: tetrahydrofuran. | |
{kind=link}
The above results showed that easily accessible phosphite and phosphoramidite ligands could compete with chiral phosphine ligands in asymmetric hydrogenation of unsaturated substrates.Lyubimov et al. further performed a Ir-catalyzed asymmetric hydrogenation of 2-methylindole 32a using phosphoramidite 43 or 44 as ligand and I2 as additive, delivering the product (R)-8a in quantitative conversion with good enantioselectivity (Scheme 13) [24].

|
Download:
|
| Scheme 13. Ir-catalyzed asymmetric hydrogenation of 2-methylindole using phosphoramidite ligand and I2 additive. | |
{kind=link}
Cationic Ru/chiral diamine complexes are very efficient catalysts for the asymmetric hydrogenation of several N-heteroaromatic compounds and ketimines with excellent enantioselectivity [25]. The superiority of these catalysts were demonstrated by Fan and co-workers, who established a highly enantioselective synthesis of indolines by asymmetric hydrogenation of unprotected 1H-indoles 7 (Scheme 14a) [14]. This protocol featured broad substrate scope, excellent enantioselectivity and diastereoselectivity, mild reaction conditions (ambient temperature and pressure), and no use of additives. Further mechanism study revealed that the indole substrate was activated by the in situ generated TfOH from dihydrogen and the cationic ruthenium complex, and a Ru-H species was also formed to reduce the activated substrate 45 via a hydride transfer process.

|
Download:
|
| Scheme 14. Asymmetric hydrogenation of unprotected 1H-and 3H-indoles. | |
{kind=link}
They further expanded this catalytic system to 3H-indoles 46, leading to the synthesis of 2, 3, 3-trisubstituted indolines (R)-47 (Scheme 14b) [14]. It is worthy to mention that 3H-indoles are much more difficult substrates for hydrogenation, and transitionmetal-catalyzed asymmetric hydrogenation of this class of substrates has not been reported yet [26]. Shortly after, Rueping et al. also reported an asymmetric iridium-catalyzed hydrogenation protocol of 3H-indoles 46 with (R)-DM-Seg-Phos ligand 48 (Scheme 14c) [27].
Almost at the same time, Touge and Arai discovered that various unprotected indole compounds 7 could be efficiently hydrogenated by diamine-Ru(Ⅱ)-BF4 catalysts 23 with weakly acidic HFIP as the solvent (Scheme 15) [28]. Moreover, the solvent could be easily recovered quantitatively by simple distillation and reused in the next hydrogenation without loss of yield or enantioselectivity. The isotopic labeling experimental results proved that unprotected indole compounds were activated in the acidic HFIP solvent to form an iminium intermediate, which was then hydrogenated efficiently by the diamine-Ru(Ⅱ)-BF4 complexes 23 through cooperation interaction between ruthenium-hydride, amine-NH and iminium intermediate, enabling asymmetric indoline synthesis. CH/π interaction between a hydrogen atom on the η6-arene and the aromatic ring of a substrate was also proposed in the transition state 49.

|
Download:
|
| Scheme 15. Asymmetric hydrogenation of unprotected 1H-indoles catalyzed by Ru/chiral diamine complexes. | |
{kind=link}
3.2. Asymmetric hydrosilylation of indoles
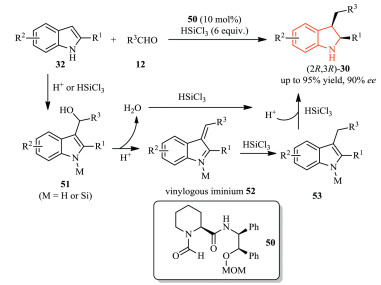
Catalytic hydrosilylation has been established as a powerful strategy for the reduction of unsaturated compounds, such as carbonyl, enamino and heteroaromatic compounds [29]. For example, works from the Sun group demonstrated that employment of the organic Lewis base catalyst 50/HSiCl3 (trichlorosilane) system allowed for the easy preparation of chiral 2, 3-disubstituted indolines 30 (Scheme 16) [30]. Starting from the simple 2-substituted indoles 32 and aldehydes 12, a tandem reaction occurred, giving the corresponding products with high yield and enantioselectivity.

|
Download:
|
| Scheme 16. Asymmetric organocatalytic tandem hydrosilylation of 2, 3-disubstituted indoles. MOM: methoxymethyl. | |
{kind=link}
A possible reaction mechanism was also proposed. The 3-(α-hydroxyalkyl)indoles 51 could be easily formed through the Friedel-Crafts reaction of the activated indole substrate 32 with aldehyde 12, which subsequently underwent dehydration to give vinylogous iminium intermediate 52 in the presence of Brønsted acid. Then, reduction of 52 with trichlorosilane led to 53. Notably, the water formed as by-product in the dehydration step could be easily consumed by trichlorosilane, which not only facilitates water elimination, but also releases hydrochloride as Brønsted acid activator for the indole intermediate in the hydrosilylation. As a result, no external Brønsted acid was needed.
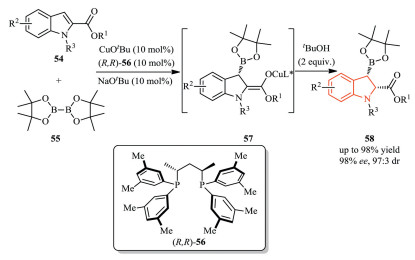
3.3. Reactions of indoles at C2 or C3 positionEnantioselective dearomatization reactions of indoles are a powerful class of transformations in the field of indole functionalization [31]. In 2015, Ito et al. realized an asymmetric borylative dearomatization of indole-2-carboxylates 54 using a chiral bisphosphine-Cu(I) catalyst and a diboron reagent 55 (Scheme 17) [32]. The experimental results and DFT calculation showed that the electron-withdrawing substituent at the 2-position in the indole substrates facilitated the addition of an active borylcopper(I) species to the indoles. Then, diastereoselective protonation of the resulting copper(I) enolate 57 by alcohol additives gave the corresponding chiral indoline derivatives 58 with excellent diastereo-and enantioselectivity.

|
Download:
|
| Scheme 17. Enantioselective borylative dearomatization of 2-substituted indoles by Cu(I) catalysis. | |
{kind=link}
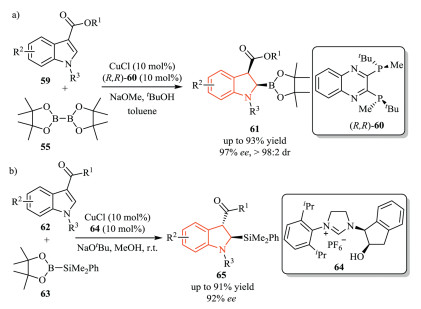
In addition to 2-substituted indoles, Xu and co-workers developed an asymmetric copper(I)-catalyzed dearomative borylation of 3-substituted indoles 59, affording highly functionalized 2, 3, 3-trisubstituted chiral indolines 61 with generally excellent yield, diastereo-and enantioselectivity (Scheme 18a) [33].Subsequently, they extended this strategy to an asymmetric dearomative silylation of 3-substituted indoles 62 (Scheme 18b) [34]. In this transformation, chiral NHC (N-heterocyclic carbene)-copper(I) complex was chosen as the catalyst instead of using phosphine ligand, which was probably due to the competitive decomposition of the silylation reagent PhMe2Si-Bpin. Mechanistic studies suggested that facile epimerization of the 3-acyl group at C3-position was responsible for the observed high diastereoselectivity.

|
Download:
|
| Scheme 18. Enantioselective borylative and silylative dearomatization of 3-substituted indoles through Cu(I) catalysis. | |
{kind=link}
In 2017, the Ready group reported an enantioselective threecomponent coupling of indoles 66, boronic esters 67 and allylic acetate 68 to provide chiral indolines 72 with high diastereoselectivity and enantioselectivity (Scheme 19a) [35]. The reaction involved the formation of boronate intermediates 70 from boronic esters and 2-lithiated indoles, outer sphere addition of this intermediate to a Pd(Ⅱ) (π-allyl) complex, and protodeborylation. In an extension of this concept, they further found that unsubstitued allyl acetate was also applicable for the synthesis of indoline boronic esters 77 using phosphoramidite 76 in asymmetric Pd-catalyzed reaction for the first time [36]. Several boronic esters were well incorporated, and selected ones were treated for the prodeborylation. Enantioselectivity was maintained at high level, while the diastereoselectivity was somewhat decreased in certain cases.

|
Download:
|
| Scheme 19. Pd-catalyzed asymmetric three-component coupling of boronic esters, indoles and allylic acetates. | |
{kind=link}
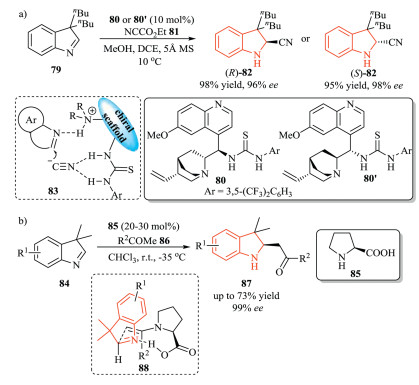
3H-Indoles have also been used in several transformations, such as hydrogenation mentioned above, nucleophilic additions, DielsAlder (DA) reactions, which provide a direct and efficient access to chiral 2-substituted indolines [14]. In 2012, Tian et al. described a qunidine-based thiourea 80-catalyzed enantioselective Strecker reaction of 3H-indoles 79 with ethyl cyanoformate 81 as a CN source, giving 2-cyanoindoline 82 with excellent yield and enantioselectivity (Scheme 20a) [37]. Both two enantiomers of the products could be obtained by using catalyst 80 and its pseudoenantiomer 80', respectively. In this process, HCN was formed as the actual cyanating agent. The high enantioselectivity might be attributed to the simultaneous activation of both indole substrate and the cyanide anion by the qunidine-based thiourea catalyst. The same group further expanded this organocatalytic strategy to the Mannich reaction of 3H-indoles 84 with ketone 86.Through this transformation, a range of optically active 2-acylmethylindolines 87 were prepared in good yield with excellent ee value. However, the scope of the ketone substrates was limited. Less active ketones such as acetophenone and cyclohexanone were not suitable for this reaction. A possible chair-like transition state model 88 was proposed, in which hydrogen-bond interaction between enamine and 3H-indole could rationalize the asymmetric induction (Scheme 20b) [38].

|
Download:
|
| Scheme 20. Organocatalytic transformations of 3H-indoles. DCE: 1, 2-dichloroethane. | |
{kind=link}
4. De novo construction of chiral 2-substituted indolines
In addition to indole functionalization, the de novo construction is an alternative efficient method to access enantioenriched 2-or 2, 3-disubstituted indolines from versatile starting materials. Most of the methods are based on the construction of pyrrolidine ring, which avoid the use of preformed indole substrates. In this section, chiral synthons strategy, organocatalysis and metal catalysis are briefly discussed.
4.1. Chiral synthons strategyIn 2013, Xiao and co-workers reported an interesting [4 + 1] annulation of chiral sulfur ylides 89 derived from (R)-BINOL and N-(ortho-chloromethyl)aryl amides 90 to access chiral 2-substituted indolines in moderate to high yield and enantioselectivity (up to 93% yield, 91% ee) (Scheme 21) [39]. Significantly, after the completion of the reaction, chiral sulfide 92 could be recovered in 95% yield, and reused in the preparation of chiral sulfur ylide.

|
Download:
|
| Scheme 21. Formal [4 + 1] annulation of chiral sulfur ylides and N-(ortho-chloromethyl)aryl amides. | |
{kind=link}
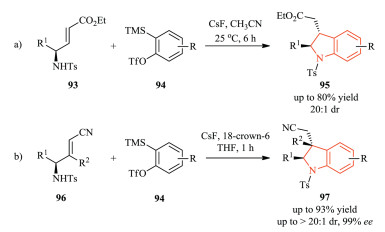
Besides [4 + 1] annulation strategy [40], [3 + 2] cycloaddition is also an effective protocol for the construction of the pyrrole ring in the indoline unit. In 2017, Sudalai et al. demonstrated a one-step formal [3 + 2] cycloaddition of in situ-generated arynes as twoatom component and γ-amino-α, β-unsaturated esters that were easily accessible from α-amino acids (Scheme 22a) [41]. This methodology featured readily accessible starting materials, mild reaction conditions, good yield and excellent level of regio-and diastereoselectivity, leading to the synthesis of 2, 3-disubstituted indolines with good yield and diastereoselectivity. The Ikawa group also reported a similar work almost at the same time.Notably, substrates with electron-withdrawing cyano group could lead to retention of the optical integrity of the arynophile, thus allowing its diverse further transformations into other functional groups (Scheme 22b) [42].

|
Download:
|
| Scheme 22. Formal [3 + 2] annulation of γ-amino-α, β-unsaturated alkenes and arynes. | |
{kind=link}
In 2018, Chang and co-workers described an intramolecular Buchwald-Hartwig cross coupling of methyl (R)-2-amino-3-(2-bromoaryl)propanoates 99, prepared from Williams intermediate 98, for the synthesis of enantiopure 2-substituted indolines 100 in excellent yield (Scheme 23) [43]

|
Download:
|
| Scheme 23. Synthesis of enantiopure 2-substituted indolines through BuchwaldHartwig cross-coupling. | |
{kind=link}
4.2. Organocatalysis strategy
Asymmetric organocatalysis has witnessed a significantly explosive growth over the past decades [44]. The development of new and efficient processes to synthesize chiral indolines via asymmetric organocatalysis has also become and will continue to be an intriguing research topic in recent years. In 2013, Asano and Matsubara demonstrated a novel method for the asymmetric synthesis of a variety of 2-substituted indolines 103 via intramolecular aza-Michael addition approach using a bifunctional aminothiourea catalyst 102. The reaction was applicable to a wide range of different substrates mediated by activation via hydrogen bonding (Scheme 24a) [45]. In 2017, Kim et al. also developed an organocatalytic intramolecular Michael addition reaction of (E)-3-(2-(2-oxopropylamino)aryl)-1-arylprop-2-en-1-ones 104a, employing a primary amine catalyst 105 derived from cinchona alkaloid (Scheme 24b) [46]. The corresponding cis-2, 3-disubstituted indoline derivatives 107a were obtained in high yield with excellent enantioselectivity and moderate diastereoselectivity. Moreover, the organocatalytic reaction of (E)-3-(2-(2-oxopropylamino)aryl)-1-alkylprop-2-en-1-ones 104b also worked quite well, delivering trans-2, 3-disubstituted indoline derivatives 107b with excellent results. As illustrated in transition state 108, the Siface of the enamine genertated from the substrate and catalyst approached the Re-face of tert-butyl α, β-unsaturated ketone moiety, forming the final S, S-configured trans-indoline 107b.

|
Download:
|
| Scheme 24. Organocatalytic intramolecular Michael addition reaction for synthesis of chiral indolines. | |
{kind=link}
Enantioselective halogenations of alkenes have recently received much attention from the synthetic community [47]. In 2014, the Wirth group reported an organocatalytic stereoselective iodoamination reaction of alkenes 109 with NIS (N-iodosuccinimide) 111 as the electrophilic iodine source using a chiral thiohydantoin catalyst 110. The reaction proceeded through halogen bonding interaction between the Lewis-basic sulfur center and the electrophilic iodine atom (Scheme 25a). On the basis of the experimental results and NMR studies, it was suggested that the additives and the electronic properties of the aromatic ring in the substrate could influence the regioselectivity of the cyclization step [48].

|
Download:
|
| Scheme 25. Organocatalytic stereoselective haloaminocyclization for synthesis of chiral indolines. | |
{kind=link}
In 2017, Deng and co-workers achieved an enantioselective bromoamination of allyl anilines 114 with NBS (N-bromosuccinimide) 116 with BINOL-derived monosulfide 115 as catalyst. This protocol provided a series of chiral 2-bromomethyl indolines 117 in good to excellent yield with high enantioselectivity (Scheme 25b).Importantly, the products could be easily transformed into other useful building blocks [49].
4.3. Metal catalysis 4.3.1. Cu(Ⅱ)-catalyzed enantioselective alkene amination reactionsThe Chemler group developed a series of Cu(Ⅱ)-catalyzed enantioselective alkene amination reactions toward C-N bond formation. They firstly reported an enantioselective intramolecular alkene aminooxygenation of γ-aminoalkenes 118 using (2, 2, 6, 6-tetramethylpiperidin-1-yl)oxyl radical (TEMPO) 120 as the oxygen source for the synthesis of chiral indoline derivatives 121 [50a].The N-O bond of the products could be reduced to the corresponding alcohols without loss of enantiopurity. The reaction could also be performed on a multigram scale [50b]. In addition, mechanistic analysis supported that the reaction occurred through an enantioselective cis aminocupration of the γ-aminoalkene 118a followed by C-Cu(Ⅱ) homolysis, thereby generating a chiral β-aminoalkyl radical 124a, which was trapped with TEMPO to afford the aminooxygenation product 121a (Scheme 26a).

|
Download:
|
| Scheme 26. Cu(Ⅱ)-catalyzed enantioselective alkene amination reactions for synthesis of chiral indoline derivatives. | |
{kind=link}
The generated radical 124a could also be intercepted by other radical acceptors, affording the corresponding difunctionalized products. In 2012, they described an enantioselective intramolecular alkene amination/intermolecular Heck-type coupling cascade process. The addition of the generated chiral carbon radical 124 to another vinyl arene 125 and subsequent oxidation provided the functionalized chiral indolines 127 (Scheme 26b) [50c]. They further developed an enantioselective alkene aminohalogenation/cyclization based on atom transfer of the generated chiral carbon radical 124 to give a wide range of chiral 2-halomethyl indolines 129 with good to excellent yield and enantioselectivity (Scheme 26c) [50d]. Based on this previous work, they reasoned that a hydroamination product could be obtained by atom transfer via hydrogen abstraction from a hydrogen donor, and thus developed a successful implementation of this strategy using 1, 4-cyclohexadiene 130 as a hydrogen source (Scheme 26d) [50e].In 2014, they also expanded the scope of copper-catalyzed inter/intramolecular alkene diamination protocol for the synthesis of 2-aminomethyl indolines 133 (Scheme 26e) [50f].
4.3.2. Pd-catalyzed C(sp3)-H bond activation/cyclizationPd-catalyzed C(sp3)-H bond activation/cyclization has been established as one of the most efficient methods in the field of heterocycle synthesis [51]. In 2012, Cramer and co-workers disclosed a Pd-catalyzed C(sp3)-H bond activation/cyclization of aryl triflates 134 to form indolines 137 in a highly enantioselective fashion using modular monodentate phosphine ligands 135 (Scheme 27). It was found that the ligand and carboxylic acid 136 were necessary for the metalation and played an important synergistic role in the reaction [52].

|
Download:
|
| Scheme 27. Pd-catalyzed C(sp3)-H bond activation toward synthesis of chiral indoline derivatives. | |
{kind=link}
In the same year, the Kündig group reported a Pd-catalyzed C (sp3)-H/C(Ar) coupling of racemic carbamates 138 with chiral NHC ligands 139. 2-Methylindolines 141 were obtained with high enantioselectivity using PCy3HBF4, IPrHCl and (R, R)-Me-DUPHOS as the chiral ligand. However, when (S, S)-139a, (S, S)-139b were used, regiodivergent reactions of racemic mixtures occurred to yield enantioenriched 2-substituted and 2, 3-disubstituted indolines via asymmetric methyl C-H activation or methylene C-H activation (Scheme 28) [53]. The combined theoretical and experimental studies showed that the Pd-NHC catalyzed C(sp3)-H arylation proceeds via a concerted metalation-deprotonation (CMD) pathway. The stereoselectivity was determined by this CMD step [54]. In 2014, they continued to probe the scope and limitations of this transformation, and found that single regioisomeric, enantiopure products rather than mixtures were produced with enantiopure starting materials. Based on the DFT study, the reactivity of methyl versus methylene groups was analyzed for the rationalization of the selectivity observed in regiodivergent reaction of a racemic mixture [55].

|
Download:
|
| Scheme 28. Pd-catalyzed C(sp3)-H/C(Ar) coupling of racemic carbamates with chiral NHC ligands. | |
{kind=link}
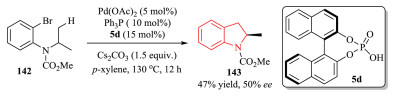
In 2016, Duan et al. reported Pd-catalyzed asymmetric C(sp3)-H activation of N-aryl carbamate 142 with chiral phosphoric acid 5d.After condition optimizations, 2-methylindolines 143 were obtained with moderate yield and enantioselectivity (Scheme 29) [56].

|
Download:
|
| Scheme 29. Pd-catalyzed C(sp3)-H activation reaction of N-aryl carbamates with chiral phosphoric acid. | |
{kind=link}
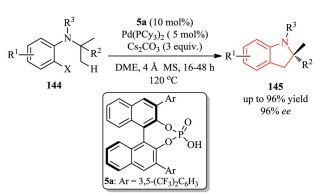
Meanwhile, the Baudoin group developed a Pd-catalyzed enantioselective C(sp3)–H activation/cyclization reaction of 144 using a BINOL-derived phosphoric acid 5a and an achiral phosphine ligand (Scheme 30). High levels of enantioselectivity for a variety of indoline products 145 were achieved by fine tuning phosphoric acid pre-catalyst, and other reaction parameters (solvents, additives etc.) [57].

|
Download:
|
| Scheme 30. Pd-catalyzed C(sp3)-H activation/cyclization reaction of carbamates in the presence of chiral phosphoric acid. DME: 1, 2-dimethoxyethane. | |
{kind=link}
4.3.3. Decarboxylation/cycloaddition sequence
1, 4-Dipoles, generated by transition-metal catalysis from readily available substrates, were found to be versatile intermediate in the field of cycloaddition reactions [58]. In 2014, the group of Lu and Xiao developed a Pd-catalyzed asymmetric decarboxylation/cycloaddition sequence of vinyl benzoxazinanones 146 as 1, 4-dipole precursors and sulfur ylides 147 (Scheme 31) [59]. A wide range of trans 2-acyl-3-vinyl indolines 149 were synthesized in high yield with excellent diastereo-and enantioselectivity by using the chiral phosphoramidite ligand 148. Notably, the Pdstabilized zwitterionic intermediates 150 were firstly trapped with nucleophilic dipolarophiles (sulfur ylides) and found to be crucial for controlling the branched selectivity.

|
Download:
|
| Scheme 31. Pd-catalyzed asymmetric [4 + 1] cycloaddition reactions of vinyl benzoxazinones with sulfur ylides. | |
{kind=link}
In 2016, they further expanded the application of vinyl benzoxazinanone 146a to the synthesis of chiral indolines 154 via the combination of two Pd-catalyzed sequential reactions (decarboxylative amination and haloamination) in one pot with good synthetic efficiency and excellent enantiocontrol (Scheme 32) [60].

|
Download:
|
| Scheme 32. Pd-catalyzed asymmetric allylic amination reaction and subsequent one-pot cyclization. | |
{kind=link}
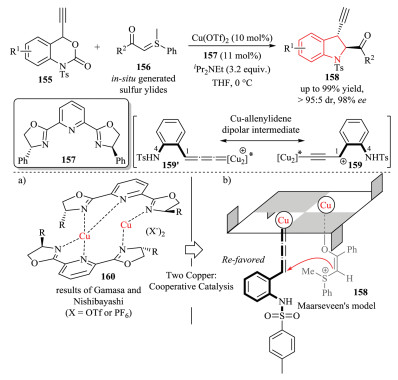
Soon after, they designed and synthesized ethynyl benzoxazinones 155, which showed similar reactivity as compared to vinyl benzoxazinanones 146. They found that the Cu-catalyzed decarboxylative asymmetric [4 + 1] cycloaddition reactions of ethynyl benzoxazinones 155 with sulfur ylides 156 also proceeded smoothly using PyBOX ligand 157 (Scheme 33) [61]. The reaction produced 3-ethynyl-substituted indolines 158 in high yield with excellent asymmetric induction. Remarkably, based on the experimental results of non-linear effects, they suggested that a cooperative bi-Cu complex 160 participated in the enantiodetermining step of the reaction. As depicted in the proposed model 161, the orientation of sulfur ylides was governed by one copper. Thus the sulfur ylide approached the Re-face of the Cu-allenylidene intermediate 159', which was formed from the ethynyl benzoxazinon 155 and another copper in the decarboxylation step, acting as the reactive 1, 4-dipole. Notably, the use of in situ-generated sulfur ylides by deprotonation of the corresponding sulfonium salt with an excess of iPr2NEt dramatically improved the selectivity.

|
Download:
|
| Scheme 33. Cu-catalyzed decarboxylative asymmetric [4 + 1] cycloaddition reactions of ethynyl benzoxazinones with sulfur ylides using PyBOX ligand. | |
{kind=link}
4.3.4. Intramolecular capture of organocuprate intermediates with imines as electrophiles
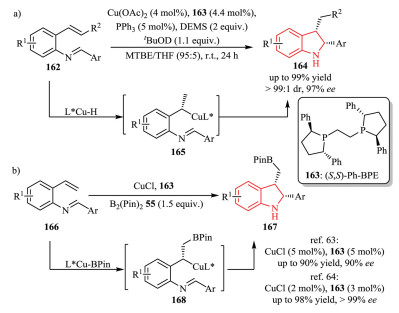
In 2015, the Buchwald group reported a Cu-catalyzed intramolecular cyclization of various vinyl arenes 162 with an imine moiety at the ortho-position (Scheme 34a) [62]. This protocol allowed for the synthesis of 2, 3-disubstituted indolines 164 with high diastereo-and enantioselectivity, featuring mild reaction conditions and high functional group tolerance. It was postulated that the reaction proceeded via a CuH-catalyzed Markovnikov addition of styrenes, thus forming organocuprate intermediate 165 and subsequent intramolecular trapping with tethered imine electrophiles. The active L*CuH species could be formed by reacting Cu(OAc)2 with a chiral ligand 163 and a stoichiometric amount of the hydrosilane DEMS (diethoxymethylsilane).

|
Download:
|
| Scheme 34. Cu-catalyzed strategy of various vinyl arenes with an imine moiety at the ortho-position for the synthesis of 2, 3-disubstituted indolines. MTBE: methyl tert-butyl ether. | |
{kind=link}
In 2018, the Yun group [63] and Xiong group [64] independently developed a Cu-catalyzed enantioselective intramolecular borylative cyclization of the same starting material with B2(Pin)2 55 at the same time. In this process, the in situ generated L*Cu-BPin catalyst played the same role as L*CuH species mentioned above.As a result, a wide range of enantioenriched 2, 3-disubstituted indolines bearing the versatile BPin functional group 167 were obtained in a highly regio-, diastereo-, and enantioselective manner by single operation (Scheme 34b).
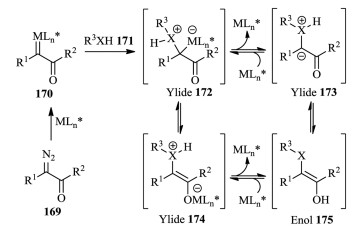
4.3.5. Metal carbenesCarbenes have also been well established as versatile intermediates in organic synthesis, and aroused much research interest.Thus, the versatile reactivity modes of carbenes, especially the metal carbenes, rendered them to be one of the most significant and useful intermediates in the past decades [65]. Diazo compounds are commonly used as carbene precursors [66].Asymmetric catalytic cascade reactions involving trapping of the active ylides 172, generated from the reaction of diazo compounds 169 with a nucleophile 171, could enable rapid and efficient assembly of complex molecules (Scheme 35) [67].

|
Download:
|
| Scheme 35. Formation of the active ylides from the reaction of diazo compounds with a nucleophile. | |
{kind=link}
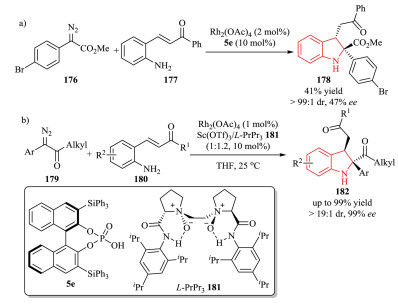
For example, Hu et al. disclosed a highly diastereoselective intramolecular trapping of ammonium ylides with enones 177 through a Rh(Ⅱ)/Brønsted acid cooperative catalytic system. Chiral BINOL-derived phosphoric acid 5e was used for the asymmetric synthesis of 2, 2, 3-trisubstituted indoline 178 with 47% ee (Scheme 36a) [68]. In 2018, Liu, Feng and co-workers reported a Rh(Ⅱ)/chiral N.N'-dioxide-Sc(Ⅲ) complex bimetallic relay catalytic system for the reaction of α-diazoketones 179 with enones 180.The use of α-diazoketones improved the reactivity and enantioselectivity with respect to α-diazoesters. A series of chiral 2, 2, 3-trisubstituted indolines 182 were constructed with high yield (up to 99% yield), high enantioselectivity (up to 99% ee), and excellent diastereoselectivity (up to > 19:1 dr) (Scheme 36b) [69].

|
Download:
|
| Scheme 36. Intramolecular trapping of ammonium ylides with enones for synthesis of chiral 2, 2, 3-trisubstituted indolines by cooperative catalytic systems. | |
{kind=link}
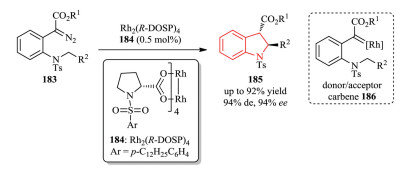
The metal-catalyzed C-H insertion of diazo compounds has been widely investigated for C-C bond formation and also applicable for the synthesis of diverse heterocycles [66]. In 2017, the Wirth group reported an efficient enantioselective synthesis of trans-2, 3-disubstituted indolines 185 with good yield and enantioselectivity through Rh(Ⅱ) catalyzed C-H insertion from α-diazo carbonyl compounds 183 (Scheme 37) [70]. In this process, donor/acceptor carbene 186 was involved.

|
Download:
|
| Scheme 37. Chiral dirhodium-catalyzed C-H insertion of α-diazocarbonyl compounds. | |
{kind=link}
In 2018, Doyle, Xu and their co-workers disclosed that a novel chiral dirhodium complex 189 catalyzed an asymmetric cascade carbene/alkyne metathesis (CAM) reaction of propargyl diazoacetates 187 to give intermediate 188 (Scheme 38) [71]. Then, highly site-selective intramolecular C(sp3)-H bond insertion provided a general access to chiral indolines in high yield with excellent enantiocontrol. Experimental and DFT studies suggested that the regioselectivity was determined by the competition between electronically favored carbocation stability and sterically demanding catalyst and substrates. The chiral dirhodium catalysts promoted both the carbene/alkyne metathesis process and the observed asymmetric induction of the terminating C-H insertion reaction. The donor/donor carbene intermediate 188 was generated in situ via carbene/alkyne metathesis with reduced reactivity compared to donor/acceptor carbene, which would enable high site-selectivity and enantioselectivity.

|
Download:
|
| Scheme 38. Chiral dirhodium-catalyzed asymmetric cascade CAM reaction /intramolecular C(sp3)-H bond insertion. | |
{kind=link}
Almost at the same time, the Shaw group expanded the C-H insertion protocol of donor/donor carbene to the asymmetric synthesis of indolines. Oxidation of hydrazones 194 provided the corresponding requisite diazo precursor in situ for donor/donor carbene, which avoided the potential explosiveness and practical problems associated with diazo compounds (Scheme 39). They also studied the impact of sterics and electronics in C-H insertion of donor/donor carbene on the regiochemistry [72].

|
Download:
|
| Scheme 39. Chiral dirhodium-catalyzed C-H insertion reaction of donor/donor carbene from hydrazones. | |
{kind=link}
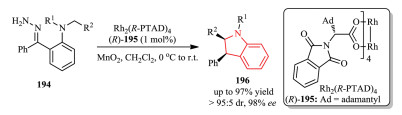
Zhang, Zhu, and co-workers continued their efforts to develop more safe and practical carbene chemistry based on enynones 197 as efficient carbene precursors for intramolecular C-H insertion (Scheme 40) [73]. The reaction gave rise to a range of enantioenriched indolines 199 in a one-pot manner and slow addition of reagent was not necessary.

|
Download:
|
| Scheme 40. Chiral dirhodium-catalyzed C-H insertion of donor/donor carbene by a nondiazo approach. | |
{kind=link}
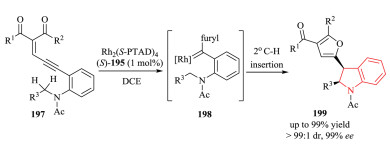
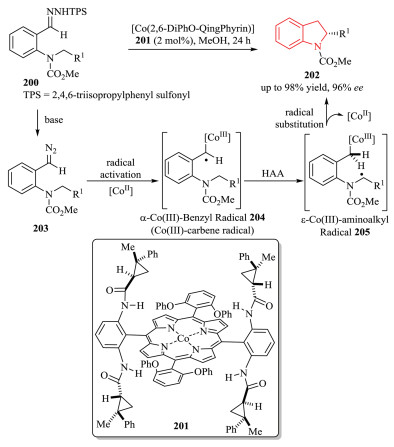
Radical chemistry offers a potentially powerful approach for the construction of organic molecules with unique reactivity and selectivity, which are complementary to the ionic chemistry.However, the control of stereoselectivity, particularly the enantioselectivity of radical reactions is typically difficult. To solve the inherent disadvantages of radical reactions, a more promising trend has been moved to metalloradical catalysis (MRC), which has emerged as a conceptually new approach involving the catalytic generation of metal-centered radical for the development of stereoselective radical processes [74]. In 2018, Zhang and coworkers developed a novel Co(Ⅱ)-based metalloradical system for enantioselective radical alkylation of C(sp3)-H bonds towards the synthesis of chiral indolines 202 in high yield with excellent enantioselectivity (Scheme 41) [75]. Using the new catalyst 2, 6-DiPhO-QingPhyrin 201, the Co(Ⅱ)-catalyzed system enables the activation of in situ generated ortho-aminoaryldiazomethanes 203 as radical precursors at room temperature. The in situ-formed α-Co(Ⅲ)-benzyl radical intermediate 204 (also known a Co(Ⅲ)-carbene radical) underwent intramolecular hydrogen atom abstraction (HAA) of C-H bond, followed by 5-exo-tet radical cyclization. Donor-type carbene was firstly involved in the asymmetric construction of 2-substituted indolines 202 through C2(sp3)-C3(sp3) bond formation. Mechanistic studies were conducted to provide evidences for the stepwise radical pathway.

|
Download:
|
| Scheme 41. Enantioselective synthesis of chiral indolines via radical C-H alkylation by Co(Ⅱ)-MRC. | |
{kind=link}
4.3.6. Ullmann C-N coupling
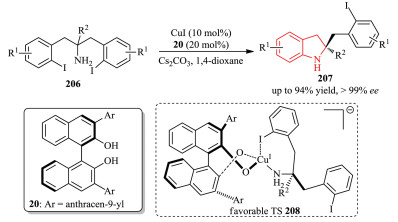
Ullmann C-N Coupling has offered a classic and powerful method for C-N bond formation [76]. In this context, the Cai group has devoted a great deal of efforts towards developing asymmetric aryl C-N couplings by asymmetric desymmetrization strategy [77].In 2012, they developed an asymmetric Cu-catalyzed desymmetric intramolecular Ullmann C-N coupling reaction of 1, 3-bis(2-iodoaryl)propan-2-amines 206, which led to the enantioselective preparation of indolines 207 in both high yield and ee value (Scheme 42) [78]. The transition state 208 for the chirality induction was proposed. The favorable model implied that the deformation of the substrate and the oxygen-copper interaction attribute to the excellent enantioselectivity.

|
Download:
|
| Scheme 42. Cu(I)-catalyzed desymmetric intramolecular Ullmann C-N coupling reaction of 1, 3-bis(2-iodoaryl)propan-2-amines. | |
{kind=link}
5. Conclusion and outlook
In this review, a number of recent approaches to the construction of chiral 2-substituted indolines were described.Among these reported examples, the indole functionalization and metal-catalyzed de novo construction were the most straightforward and widely used methodologies. It goes without saying that this research field has emerged as one of the fast growing fields in heterocycle synthesis, and will continue to be flourishing in the future. We hope that this review will provide researchers a comprehensive updated overview of the efforts devoted to assembly of chiral 2-substituted indolines. It is likely that more and more powerful and mild approaches to enantioenriched 2-substituted indolines will thus be developed. In addition, it can be anticipated that there will be more versatile synthetic transformations of optically active 2-substituted indolines to other more complex molecules such as natural products or pharmaceuticals in the near future.
AcknowledgmentsWe thank the National Natural Science Foundation of China (Nos. 21702121, 91856119 and 21622201), the Science and Technology Department of Hubei Province (Nos. 2016CFA050 and 2017AHB047), and the Program of Introducing Talents of Discipline to Universities of China (111 Program, No. B17019) for support of our research in this area.
| [1] |
(a) J. Dunlop, K.L. Marquis, H.K. Lim, et al., CNS Drug Rev. 12 (2006) 167-177; (b) D. Crich, A. Banerjee, Acc. Chem. Res. 40 (2007) 151-161; (c) D. Zhang, H. Song, Y. Qin, Acc. Chem. Res. 44 (2011) 447-457. |
| [2] |
F. Andersson, E. Hedenström, Tetrahedron: Asymmetry 17 (2006) 1952-1957. DOI:10.1016/j.tetasy.2006.07.002 |
| [3] |
R. Neelamegam, T. Hellenbrand, F.A. Schroeder, et al., J. Med. Chem. 57 (2014) 1488-1494. DOI:10.1021/jm401802f |
| [4] |
(a) L. Xiang, D. Xing, W. Wang, et al., Phytochemistry 66 (2005) 2595-2601; (b) W.G. Kim, J.P. Kim, H. Koshino, et al., Tetrahedron 53 (1997) 4309-4316; (c) A. Rakhit, M.E. Hurley, V. Tipnis, et al., J.Clin. Pharmacol. 26 (1986) 156-164; (d) S. Adachi, K. Koike, I. Takayanagi, Pharmacology 53 (1996) 250-258; (e) V. Certal, J.C. Carry, F. Halley, et al., J. Med. Chem. 57 (2014) 903-920; (f) J. Bermudez, S. Dabbs, K.A. Joiner, et al., J. Med. Chem. 33 (1990) 1929-1932. |
| [5] |
(a) S. Anas, H.B. Kagan, Tetrahedron: Asymmetry 20 (2009) 2193-2199; (b) D. Liu, G. Zhao, L. Xiang, Eur. J. Org. Chem. 2010 (2010) 3975-3984; (c) B. Zhang, W. Qin, Y. Duan, et al., Chin. J. Org. Chem. 32 (2012) 1359-1367. |
| [6] |
(a) F.F. Huerta, A.B.E. Minidis, J.E. Bäckvall, Chem. Soc. Rev. 30 (2001) 321-331; (b) E. Vedejs, M. Jure, Angew. Chem. Int. Ed. 44 (2005) 3974-4001; (c) E. Fogassy, M. Nógrádi, D. Kozma, et al., Org. Biomol. Chem. 4 (2006) 3011-3030; (d) H. Pellissier, Adv. Synth. Catal. 353 (2011) 1613-1666; (e) V.P. Krasnov, D.A. Gruzdev, G.L. Levit, Eur. J. Org. Chem. 2012 (2012) 1471-1493. |
| [7] |
M. Breuer, K. Ditrich, T. Habicher, et al., Angew. Chem. Int. Ed. 43 (2004) 788-824. DOI:10.1002/anie.200300599 |
| [8] |
M. López-Iglesias, E. Busto, V. Gotor-Fernández, et al., J. Org. Chem. 77 (2012) 8049-8055. DOI:10.1021/jo301307q |
| [9] |
(a) I. Čorić, S. Müeller, B. List, J. Am. Chem. Soc. 132 (2010) 17370-17373; (b) I. Čorić, B. List, Nature 483 (2012) 315-319; (c) I. Čorić, J.H. Kim, T. Vlaar, et al., Angew. Chem. Int. Ed. 52 (2013) 3490-3493; (d) J.H. Kim, I. Čorić, B. List, et al., J. Am. Chem. Soc. 137 (2015) 1778-1781. |
| [10] |
(a) K. Saito, M. Yamanaka, T. Akiyama, et al., J. Am. Chem. Soc. 135 (2013) 11740-11743; (b) K. Saito, T. Akiyama, Angew. Chem. Int. Ed. 55 (2016) 3148-3152. |
| [11] |
Y. Wang, G. Li, G. Zhao, et al., Tetrahedron Lett. 58 (2017) 2993-2996. DOI:10.1016/j.tetlet.2017.06.057 |
| [12] |
J.I. Murray, N.J. Flodén, A.C. Spivey, et al., Angew. Chem. Int. Ed. 56 (2017) 5760-5764. DOI:10.1002/anie.201700977 |
| [13] |
W. Yang, Y. Zeng, Q. Cai, et al., Org. Lett. 15 (2013) 3598-3601. DOI:10.1021/ol401449b |
| [14] |
Z. Yang, F. Chen, Q.H. Fan, et al., Angew. Chem. Int. Ed. 55 (2016) 13863-13866. DOI:10.1002/anie.201607890 |
| [15] |
Z. Luo, L. Zhang, Z. Wang, et al., Chem. Commun. 54 (2018) 13503-13506. DOI:10.1039/C8CC07336H |
| [16] |
Q.A. Chen, Z.S. Ye, Y.G. Zhou, et al., Chem. Soc. Rev. 42 (2013) 497-511. DOI:10.1039/C2CS35333D |
| [17] |
Y. Duan, C.B. Yu, Y.G. Zhou, et al., Org. Biomol. Chem. 10 (2012) 1235-1238. DOI:10.1039/C1OB06777J |
| [18] |
Y. Duan, H.-J. Fan, Y.-G. Zhou, et al., J. Am. Chem. Soc. 136 (2014) 7688-7700. DOI:10.1021/ja502020b |
| [19] |
D.Y. Zhang, C.B. Yu, Y.G. Zhou, et al., Tetrahedron Lett. 53 (2012) 2556-2559. DOI:10.1016/j.tetlet.2012.03.036 |
| [20] |
C. Li, Y. Liu, W. Zhang, et al., Tetrahedron 69 (2013) 6839-6844. DOI:10.1016/j.tet.2013.06.016 |
| [21] |
C.B. Yu, J. Wang, Y.G. Zhou, Org. Chem. Front. 5 (2018) 2805-2809. DOI:10.1039/C8QO00710A |
| [22] |
J. Wen, L.W. Chung, X Zhang, et al., Org. Lett. 20 (2018) 2143-2147. DOI:10.1021/acs.orglett.8b00312 |
| [23] |
J.L. Núñez-Rico, H. Fernández-Pérez, A. Vidal-Ferran, Green Chem. 16 (2014) 1153-1157. DOI:10.1039/c3gc42132e |
| [24] |
(a) S.E. Lyubimov, D.V. Ozolin, V.A. Davankov, Tetrahedron Lett. 55 (2014) 3613-3614; (b) D.V. Ozolin, S.E. Lyubimov, V.A. Davankov, Russ. Chem. Bull. 63 (2014) 2399-2401. |
| [25] |
(a) D.S. Wang, Q.A. Chen, Y.G. Zhou, et al., Chem. Rev. 112 (2012) 2557-2590; (b) Z. Wu, M. Perez, V. Ratovelomanana-Vidal, et al., Angew. Chem. Int. Ed. 52 (2013) 4925-4928; (c) A. Bartoszewicz, N. Ahlsten, B. Martin-Matute, Chem. -Eur. J. 19 (2013) 7274-7302. |
| [26] |
A.M. Kluwer, A.M. van der Burg, J.N.H. Reek, et al., Adv. Synth. Catal. 354 (2012) 89-95. DOI:10.1002/adsc.201100422 |
| [27] |
R. Borrmann, N. Knop, M. Rueping, Chem. -Eur. J. 23 (2017) 798-801. DOI:10.1002/chem.201605450 |
| [28] |
T. Touge, T. Arai, J. Am. Chem. Soc. 138 (2016) 11299-11305. DOI:10.1021/jacs.6b06295 |
| [29] |
Q. Shi, Z. Chen, J. Hu, Curr. Org. Chem. 22 (2018) 557-580. DOI:10.2174/1385272822666171227145613 |
| [30] |
L. Chen, C. Wang, J. Sun, et al., Adv. Synth. Catal. 356 (2014) 2224-2230. DOI:10.1002/adsc.201301133 |
| [31] |
(a) M. Bandini, A. Eichholzer, Angew. Chem. Int. Ed. 48 (2009) 9608-9644; (b) C.X. Zhuo, W. Zhang, S.L. You, Angew. Chem. Int. Ed. 51 (2012) 12662-12686; (c) Q. Ding, X. Zhou, R. Fan, Org. Biomol. Chem. 12 (2014) 4807-4815; (d) C.X. Zhuo, C. Zheng, S.L. You, Acc. Chem. Res. 47 (2014) 2558-2573; (e) C. Zheng, S.L. You, Chem. 1 (2016) 830-857. |
| [32] |
(a) K. Kubota, K. Hayama, H. Ito, et al., Angew. Chem. Int. Ed. 54 (2015) 8809-8813; (b) K. Hayama, K. Kubota, H. Ito, et al., Chem. Lett. 46 (2017) 1800-1802. |
| [33] |
L. Chen, J.J. Shen, S. Xu, et al., Chem. Sci. 9 (2018) 5855-5859. DOI:10.1039/C8SC01815D |
| [34] |
Y. Shi, Q. Gao, S. Xu, J. Org. Chem. 83 (2018) 14758-14767. DOI:10.1021/acs.joc.8b02308 |
| [35] |
S. Panda, J.M. Ready, J. Am. Chem. Soc. 139 (2017) 6038-6041. DOI:10.1021/jacs.7b01410 |
| [36] |
S. Panda, J.M. Ready, J. Am. Chem. Soc. 140 (2018) 13242-13252. DOI:10.1021/jacs.8b06629 |
| [37] |
Y.D. Shao, S.K. Tian, Chem. Commun. 48 (2012) 4899-4901. DOI:10.1039/c2cc31001e |
| [38] |
D.J. Cheng, S.K. Tian, Adv. Synth. Catal. 355 (2013) 1715-1718. DOI:10.1002/adsc.201300161 |
| [39] |
Q.Q. Yang, L.Q. Lu, W.J. Xiao, et al., Chem. -Eur. J. 19 (2013) 8401-8404. DOI:10.1002/chem.201300988 |
| [40] |
(a) J.R. Chen, L.Q. Lu, W.J. Xiao, et al., Chem. Rev. 115 (2015) 5301-5365; (b) Q.Q. Yang, W.J. Xiao, Eur. J. Org. Chem. 2017 (2017) 233-236. |
| [41] |
R.D. Aher, G.M. Suryavanshi, A. Sudalai, J. Org. Chem. 82 (2017) 5940-5946. DOI:10.1021/acs.joc.7b00439 |
| [42] |
T. Ikawa, Y. Sumii, S. Akai, et al., Synlett 29 (2018) 530-536. DOI:10.1055/s-0036-1591722 |
| [43] |
Q. Zhang, C. Song, J. Chang, et al., Chin. J. Org. Chem. 38 (2018) 221-227. DOI:10.6023/cjoc201708002 |
| [44] |
(a) A. Berkessel, H. Gröger, Asymmetric Organocatalysis, Wiley-VCH: Meinheim, 2004; (b) P.I. Dalko, Enantioselective Organocatalysis, Wiley-VCH: Weinheim, 2007; (c) B. List, Chem. Rev. 107 (2007) 5413-5415; (d) D.W.C. MacMillan, Nature 455 (2008) 304-308; (e) L.S. Hegedus, J. Am. Chem. Soc. 131 (2009) 17995-17997; (f) A. Moyano, R. Rios, Chem. Rev. 8 (2011) 4703-4832. |
| [45] |
R. Miyaji, K. Asano, S. Matsubara, Org. Lett. 15 (2013) 3658-3661. DOI:10.1021/ol401538b |
| [46] |
J. Lee, K.M. Ko, S.G. Kim, RSC Adv. 7 (2017) 56457-56462. DOI:10.1039/C7RA10775G |
| [47] |
(a) A. Castellanos, S.P. Fletcher, Chem. -Eur. J. 17 (2011) 5766-5776; (b) S.E. Denmark, W.E. Kuester, M.T. Burk, Angew. Chem. Int. Ed. 51 (2012) 10938-10953; (c) U. Hennecke, Chem. -Eur. J. 7 (2012) 456-465; (d) C.K. Tan, Y.Y. Yeung, Chem. Commun. 49 (2013) 7985-7996; (e) K. Murai, H. Fujioka, Heterocycles 87 (2013) 763-805; (f) Y.A. Cheng, W.Z. Yu, Y.Y. Yeung, Org. Biomol. Chem. 12 (2014) 2333-2343. |
| [48] |
P. Mizar, A. Burrelli, T. Wirth, et al., Chem. -Eur. J. 20 (2014) 13113-13116. DOI:10.1002/chem.201404762 |
| [49] |
S.N. Yu, Y.L. Li, J. Deng, Adv. Synth. Catal. 359 (2017) 2499-2508. DOI:10.1002/adsc.201700106 |
| [50] |
(a) M.C. Paderes, J.B. Keister, S.R. Chemler, J. Org. Chem. 78 (2013) 506-515; (b) F.C. Sequeira, M.T. Bovino, S.R. Chemler, et al., Synthesis 44 (2012) 1481-1484; (c) T.W. Liwosz, S.R. Chemler, J. Am. Chem. Soc. 134 (2012) 2020-2023; (d) M.T. Bovino, S.R. Chemler, Angew. Chem. Int. Ed. 51 (2012) 3923-3927; (e) B.W. Turnpenny, K.L. Hyman, S.R. Chemler, Organometallics 31 (2012) 7819-7822; (f) B.W. Turnpenny, S.R. Chemler, Chem. Sci. 5 (2014) 1786-1793. |
| [51] |
(a) R. Jazzar, J. Hitce, O. Baudoin, et al., Chem. -Eur. J. 16 (2010) 2654-2672; (b) O. Baudoin, Chem. Soc. Rev. 40 (2011) 4902-4912; (c) H. Li, B.J. Li, Z.J. Shi, Catal. Sci. Technol. 1 (2011) 191-206; (d) N. Dastbaravardeh, M. Christakakou, M. Schnürch, et al., Synthesis 46 (2014) 1421-1439; (e) J.F. Hartwig, J. Am. Chem. Soc. 138 (2016) 2-24. |
| [52] |
T. Saget, S.J. Lemouzy, N. Cramer, Angew. Chem. Int. Ed. 51 (2012) 2238-2242. DOI:10.1002/anie.201108511 |
| [53] |
D. Katayev, M. Nakanishi, E.P. Kündig, et al., Chem. Sci. 3 (2012) 1422-1425. DOI:10.1039/c2sc20111a |
| [54] |
E. Larionov, M. Nakanishi, E.P. Kündig, et al., Chem. Sci. 4 (2013) 1995-2005. DOI:10.1039/c3sc00098b |
| [55] |
D. Katayev, E. Larionov, E.P. Kündig, et al., Chem. -Eur. J. 20 (2014) 15021-15030. DOI:10.1002/chem.201403985 |
| [56] |
S. Zhang, J. Lu, W.L. Duan, et al., Chin. J. Org. Chem. 36 (2016) 752-759. DOI:10.6023/cjoc201602032 |
| [57] |
L. Yang, R. Melot, O. Baudoin, et al., Chem. Sci. 8 (2017) 1344-1349. DOI:10.1039/C6SC04006C |
| [58] |
T.R. Li, W.J. Xiao, L.Q. Lu, et al., Tetrahedron Lett. 59 (2018) 1521-1530. DOI:10.1016/j.tetlet.2018.02.081 |
| [59] |
T.R. Li, F. Tan, W.J. Xiao, et al., Nat. Commun. 5 (2014) 5500-5509. DOI:10.1038/ncomms6500 |
| [60] |
Y.N. Wang, L.Q. Lu, W.J. Xiao, et al., Org. Lett. 19 (2017) 4094-4097. DOI:10.1021/acs.orglett.7b01794 |
| [61] |
Q. Wang, L.Q. Lu, W.J. Xiao, et al., J. Am. Chem. Soc. 138 (2016) 8360-8363. DOI:10.1021/jacs.6b04414 |
| [62] |
E. Ascic, S.L. Buchwald, J. Am. Chem. Soc. 137 (2015) 4666-4669. DOI:10.1021/jacs.5b02316 |
| [63] |
D. Li, J. Kim, J. Yun, et al., Chem. Asian J. 13 (2018) 2365-2368. DOI:10.1002/asia.201800121 |
| [64] |
G. Zhang, T Xiong, Q. Zhang, et al., Org. Lett. 20 (2018) 1798-1801. DOI:10.1021/acs.orglett.8b00246 |
| [65] |
(a) H.M.L. Davies, R.E.J. Beckwith, Chem. Rev. 103 (2003) 2861-2903; (b) H.M.L. Davies, D. Morton, Chem. Soc. Rev. 40 (2011) 1857-1869; (c) M.P. Doyle, R. Duffy, L. Zhou, et al., Chem. Rev. 110 (2010) 704-724; (d) M.P. Doyle, M. Ratnikov, Y. Liu, Org. Biomol. Chem. 9 (2011) 4007-4016; (e) S. Zhu, D. Zhu, L. Chen, et al., Angew. Chem. Int. Ed. 57 (2018) 12405-12409. |
| [66] |
G. Maas, Angew. Chem. Int. Ed. 48 (2009) 8186-8195. DOI:10.1002/anie.200902785 |
| [67] |
B. Xu, S.F. Zhu, Q.L. Zhou, et al., Angew. Chem. Int. Ed. 53 (2014) 3913-3916. DOI:10.1002/anie.201400236 |
| [68] |
L. Jiang, R. Xu, W. Hu, et al., J. Org. Chem. 79 (2014) 8440-8446. DOI:10.1021/jo501282h |
| [69] |
J. Yang, X. Liu, X. Feng, et al., Org. Lett. 20 (2018) 4536-4539. DOI:10.1021/acs.orglett.8b01744 |
| [70] |
M. Santi, S.T.R. Müller, T. Wirth, et al., Eur. J. Org. Chem. 2017 (2017) 1889-1893. DOI:10.1002/ejoc.201700412 |
| [71] |
K. Dong, M.P. Doyle, X. Xu, et al., ACS Catal. 8 (2018) 9543-9549. DOI:10.1021/acscatal.8b02822 |
| [72] |
L.W. Souza, R.A. Squitieri, J.T. Shaw, et al., Angew. Chem. Int. Ed. 57 (2018) 15213-15216. DOI:10.1002/anie.201809344 |
| [73] |
D. Zhu, L. Zhang, S. Zhu, et al., Angew. Chem. Int. Ed. 55 (2016) 8452-8456. DOI:10.1002/anie.201604211 |
| [74] |
(a) M.P. Doyle, Angew. Chem. Int. Ed. 48 (2009) 850-852; (b) H. Lu, X.P. Zhang, Chem. Soc. Rev. 40 (2011) 1899-1909; (c) H. Pellissier, H. Clavier, Chem. Rev. 114 (2014) 2775-2823; (d) A. Studer, D.P. Curran, Angew. Chem. Int. Ed. 55 (2016) 58-102; (e) H. Miyabe, A. Kawashima, S. Kohtani, et al., Chem. -Eur. J. 23 (2017) 6225-6236. |
| [75] |
X. Wen, Y. Wang, X.P. Zhang, Chem. Sci. 9 (2018) 5082-5086. DOI:10.1039/C8SC01476K |
| [76] |
(a) S.V. Ley, A.W. Thomas, Angew. Chem. Int. Ed. 42 (2003) 5400-5449; (b) I.P. Beletskaya, A.V. Cheprakov, Coord. Chem. Rev. 248 (2004) 2337-2364; (c) G. Evano, N. Blanchard, M. Toumi, Chem. Rev. 108 (2008) 3054-3131; (d) F. Monnier, M. Taillefer, Angew. Chem. Int. Ed. 48 (2009) 6954-6971; (e) D. Ma, Q. Cai, Acc. Chem. Soc. 41 (2008) 1450-1460; (f) D.S. Surry, S.L. Buchwald, Chem. Sci. 1 (2010) 13-31. |
| [77] |
F. Zhou, J. Liu, Q. Cai, Synlett 27 (2016) 664-675. DOI:10.1055/s-0035-1560552 |
| [78] |
(a) F. Zhou, J. Guo, Q. Cai, et al., J. Am. Chem. Soc. 134 (2012) 14326-14329; (b) J. Liu, J. Yan, Q. Cai, et al., Synthesis 46 (2014) 1917-1923. |