 2020, Vol. 31
2020, Vol. 31 


Being responsive to multiple stimuli such as heat, light, redox potential, pH or mechanical stress, polymer materials possess many unique properties that play a significant role in drug-delivery systems, molecular devices, or novel membranes [1-3]. Among these functionalities, responsive polymer which utilizes light or ultrasound as an external stimulus offers a range of advantages, including ease of application, non-invasion and controllability both spatially and temporally. Particularly, mechano-responsive polymers which could undergo various chemical changes upon force to degrade [4-6], has become an emerging field that attracted much attention. In terms of the utility of dual response for the control release of functional species and chain dissociation site-specifically in different environments, it remains important to develop new types of polymer materials that are responsive to both light and mechanical force.
On the other hand, carbon monoxide (CO), generated endogenously in mammals, akin to nitric oxide (NO) [7-9], as an important gas transmitter, plays important roles in anti-inflammatory, antihypertensive, and antiapoptotic pathways as a physiological signal molecule [10]. However, the medical use of CO gas is greatly limited due to its high toxicity at high concentrations and the lack of targeted release [11]. In general, CO-releasing molecules (CORM) based on metal carbonyl complexes are developed [12, 13] for controlled delivery of CO. For example, Wang et al. performed the host–guest interaction of cyclodextrin with air-sensitive Fe(CO)2 (Fp) pendent groups in poly(Fp-methylstyrene), which is stable in solid or solution and shows light-trigged CO releasing and antimicrobial behavior [14]. However, metal-free CO-releasing polymer has never been hitherto reported.
In this work, we introduced the organic photo-responsive diphenyl cyclopropenone(DPCP) as CORM motif both into linear and crosslinked polymer. DPCP undergoes a photodecarbonylation reaction at the function of UV radiation, which releases CO and forms the diphenylacetylene (DPA) at the same time [15]. This design provides conceptual examples of facile approaches to construct responsive metal-free polymers with the functions of CO-releasing. Herein, the linear polymer containing DPCP in chain center was prepared by atom transfer radical polymerization (ATRP) method [16], of which the mechano-labile ester bond is linked to the DPCP group (Scheme 1). As reported before, the ester bond could be cut under ultrasound to endow the polymer with activation of photoluminescent [17] or release of dye [18]. Hence, we investigate the light and ultrasound responses of the linear polymer. Furthermore, the DPCP was incorporated into crosslinked polyurethane (DPCP-PU) as bulk material to release CO. The results would provide a strategy to fabricate the CO-release polymer.

|
Download:
|
| Scheme 1. Synthesis of 2,3-bis(4-hydroxyphenyl)-2-cyclopropen-1-one (2), diphenylcyclopropenone radical polymerization initiator (Br-DPCP-Br) and the esterified diphenylcyclopropenone-centered poly(methyl acrylate) (DPCP-PMA). (ⅰ) AlCl3, DCM (dry), −50 ℃, 2 h, r.t., overnight, 51.1%; (ⅱ) BBr3, DCM (dry), 0 ℃, 2 h, r.t., overnight, 70.9%; (ⅲ) Et3N, THF (dry) and DMF (dry), r.t., overnight, 43.5%; (ⅳ) CuBr(I), PMDETA, DMSO (dry), 50 ℃, 3 h. | |
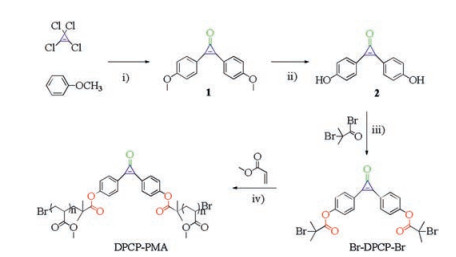{kind=link}
The synthesis and characterization details for DPCP-centered poly(methyl acrylate) (DPCP-PMA) and DPCP-PU are provided in the Supporting information. The structure of the DPCP-PMA was confirmed by high-resolution 1H NMR spectroscopy (600 MHz) (Fig. S1 in Supporting information) and the number-average molecular weight (Mn) of DPCP-PMA was 52.3 kDa with a narrow dispersity of 1.13 (Fig. S2A in Supporting information), slightly above the threshold molecular weight (Mlim, ca. 45.0 kDa) [19] making the possible daughter fragments molecular weights much lower than Mlim for the second chain scission. The control (Mn: 53.7 kDa, PDI: 1.24) with fully carbon-carbon chain was obtained (Fig. S2B in Supporting information). The crosslinked polyurethane containing DPCP group was prepared as shown in Scheme S1 (Supporting information). It can be seen from the Fig. S3 (Supporting information) that there is an intensity peak at 1843 cm−1, which was ascribed to the stretching vibration of the C=O bond in cyclopropenone. Conjugated stretching vibration of C=C and benzene ring appear at 1601 cm−1 and 1586 cm−1, which indicates that DPCP group is successfully introduced into the polyurethane.
As our expectation, the DPCP-PMA in diluted solution was fast responsive upon the irradiation of UV light, as revealed by 1H NMR. The aryl protons of DPCP unit at the chain center were two resonance peaks at 8.07 and 7.39 ppm that shifted to 7.59 and 7.15 ppm, respectively after UV irradiation. Concurrently, compared with the FT-IR spectra of small molecular diphenyl cyclopropenone before and after UV exposure (Fig. S4A in Supporting information), the FT-IR spectrum of DPCP-PMA (Fig. S4B in Supporting information) shows a clear decrease of the absorption signal of carbonyl groups (1842 cm−1) and appearance of the vibration peaks of benzene ring skeleton (1509 cm−1). A comparison of UV–vis absorption spectra of DPCP-PMA (Fig. 1A) shows the weak band around 350 nm corresponding to the S0→S1 transition (n→π*) originating from the carbonyl group disappears after UV irradiation [20]. While the UV–vis spectra of sonicated polymer were consistent with the initial, the increases in solution optical density is ascribed to the extent of chain scission [21]. These results indicated a clear photo-decarbonization reaction of DPCP-PMA, releasing CO and forming the DPA unit (C≡C) under UV irradiation [15].

|
Download:
|
| Fig. 1. (A) UV–vis absorption measurements of DPCP-PMA; (B) Photoluminescence spectroscopy (λexc =290 nm) of activation of DPCP-PMA by UV 365 nm, indicating a self-reporting CO-releasing process. | |
{kind=link}
Such chemical transformation process was also monitored by photoluminescence (PL) spectra. Fig. 1B shows the time-dependent fluorescence changes of DPCP-PMA solution. The intensity of the absorbance peak at around 350 nm grew up as irradiated time increased, indicating more amounts of DPCP unit transferred to DPA unit in a fast manner. That is, the CO-releasing process could be easily reported by the variation of the PL spectra, which is important to monitor the process in a real-time manner. The GPC trace for DPCP-PMA irradiated by UV light exhibited no chain cleavage after UV irradiation (Fig. S5 in Supporting information).
In order to quantitatively switch the DPCP moiety to DPA of DPCP-PMA on-and-off on demand, high-resolution 1H NMR spectroscopy (600 MHz) was used to trace the photo-decarbonization reaction. The DPCP-PMA solution was sealed in a transparent nuclear magnetic tube and irradiated by UV light for the set time as ON for starting decarbonization and 5 min as OFF for stopping it (Fig. 2A). The conversions of the DPCP moiety of the DPCP-PMA were calculated to be 20.0%, 32.0%, 39.8%, 43.7%, 53.2%, 61.5%, 66.2% and 75.8% for 1, 2, 3, 5, 10, 15, 20 and 25 min, respectively, based on the integral area ratio of the representative protons. Clearly, the plot of conversion with irradiation time presented an initially fast and then smooth CO-releasing characteristics. Of importance, this photo-decarbonization stopped immediately once the UV light was turned off, as shown in Fig. 2B. In contrast to the previous macromolecular and inorganic nanomaterial scaffold CORM systems [13, 22, 23], the DPCP-PMA presented a well-controlled CO releasing process without using metallic reagents.

|
Download:
|
| Fig. 2. (A) 1H NMR spectra of DPCP-PMA on UV 365 nm for different time intervals. (B) The conversion [%] of DPCP to DPA in polymer by the UV 365 nm for different time. Conversion is the integral area ratio of A7.14/(A7.14 + A7.40). | |
{kind=link}
Furthermore, the FT-TR spectrum (Fig. S6 in Supporting information) of DPCP-PU at the UV irradiation shows that the bulk crosslinked material is able to release CO. As the illumination time increases, the stretching vibration peak of the carbonyl group at 1842 cm−1 and the peaks of carbon-carbon double bonds at 1600 cm−1 and 1568 cm−1 are both gradually disappear, indicating that the DPCP in the polyurethane undergoes a photodecarbonylation reaction. After the reaction, it was observed by the naked eye that a lot of small bubbles appeared in the bulk material.
To investigate the mechanical response of linear polymers, the DPCP-PMA was subjected to sonication in dry tetrahydrofuran (THF), and aliquots were analyzed by gel permeation chromatography (GPC) over time. The GPC traces of DPCP-PMA are depicted in Fig. 3A. It is clearly observed that with the increase of sonication time, the high Mn part of the polymer decreases gradually with the emerging of a shoulder peak of which Mn is very close to the half of the initial. This trend is well consistent with the Gaussian cleavage model indicating the selective chain scission [24, 25]. Meanwhile, the control shows the molecular weight of a fully carbon-carbon chain decreased gradually but there is no shoulder or platform as a function of ultrasound time (Fig. S7 in Supporting information), which demonstrates the chain scission of pure PMA was well agreed with the random cleavage model. Hence, the GPC traces suggested that the DPCP-PMA underwent a selective chain scission.

|
Download:
|
| Fig. 3. (A) GPC traces of DPCP-PMA during ultrasound in 10 mL dry THF. (B) 1H NMR spectrum of activation of poly (methyl acrylate) chain-centered diphenylcyclopropenon by UV 365 nm, ultrasound and proton exchange experiment. (C) Plots of DI vs. sonication time of DPCP-PMA in different solvent. DI = Mn, 0 /Mn, t − 1. | |
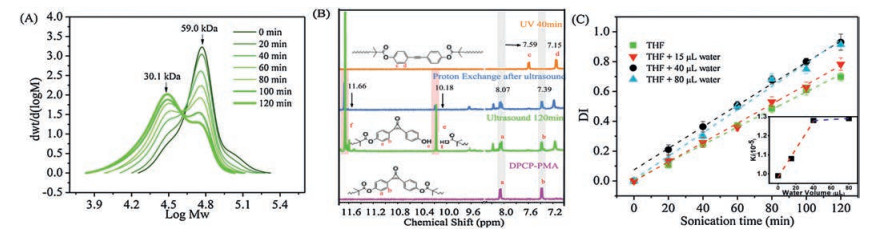{kind=link}
The DPCP moiety of PMA was completely retained after the irradiation of the ultrasound for 120 min, as revealed by the high-resolution 1H NMR spectroscopy (600 MHz) (Fig. 3B). The chemical shifts of the aromatic protons of the remained DPCP moiety of DPCP-PMA could be respectively identified to 8.07 and 7.39 ppm. No apparent changes in the chemical shifts of DPCP moiety after ultrasound were observed as compared with the pristine DPCP-PMA. It is reasonable that the cyclopropenone moiety is moderately aromatic with resonance energy in the range of 12–24 kcal/mol [26] and it has an obvious conjugated effect with two linked phenyl groups. We further discerned the end groups of the products after ultrasound using deuterium oxide (D2O) exchange NMR technique. As shown in Fig. 3B, two resonance peaks at 10.18 and 11.66 ppm that could be ascribed to the end phenol and carboxylic acid groups, respectively were clearly weakened by adding a drop of D2O. A very recent work reported that the cleavage of the phenol ester bond induced by ultrasound restored the 2-(2′-hydroxyphenyl) benzoxazole motif, which was verified by PL spectra [17]. Several previous works has suggested that the ester bonds could be mechano-chemically responsive without acknowledging the chemical structures of the products [17, 18]. Our system provided NMR evidences that confirmed the cleavage of ester bond at the chain center was a force-induced hydrolyzation reaction. When the DPCP-PMA polymer kept a free chain in dried THF system, the phenol ester bond, which has larger dipole moment than C—C bond, was easy to cleave. Therefore, it is possible to enhance the chain scission rate of the DPCP-PMA at the ester bond site by adding certain amounts of water.
To verify this idea, we next determined the rate constant for chain scission of DPCP-PMA in THF with small quantity of purposely added water. The degradation index (DI) that is a general indicator of the extent of degradation [24, 25] was used to gauge the effect of the amounts of water on the kinetic of the chain scission. The DI–t plots of the mechanochemical activation of DPCP-PMA were obtained upon adding different amounts of water. Three groups of GPC data of DPCP-PMAs exhibited pronounced changes of the molecular weights, indicating the selective chain cleavage at the chain center (Fig. S8 in Supporting information). We observed a linear correlation between DI and sonication time over the range of amount water (Fig. 3C). The rate constants of chain scission of the water volumes of 0, 15 and 40 μL were calculated to be 0.99 × 10−5, 1.08 × 10−5 and 1.28 × 10−5 min-1 (Table S1 in Supporting information). This result clearly suggests that the adding small quantity of water could promote the chain cleavage at the ester site of DPCP-PMA under ultrasound. Of surprise, for DPCP-PMA in solution with the water volume from 40 μL to 80 μL, the degradation rate constants were approximately similar (1.28 × 10−5 min-1 to 1.29 × 10−5 min-1), suggesting an up-limit for the water volume on the chain scission, probably due to the curling of PMA chains caused by water. Clearly, the precondition is that the polymer chain keeps free single chain state in a diluted solution.
In summary, we describe a facile metal-free approach to prepare CO-releasing polymer material in a switching on-off mode with temporal and spatial control. The CO-releasing process can be reported by the photoluminescence spectroscopy. The DPCP group in PMA chain central is mechanical-stable under ultrasound while the force-induced hydrolyzation at the phenol ester bond of the polymer was affected by the small amounts of exogenous water in a diluted solution. Therefore, we provided a new material with functions of site–specific CO releasing and chain scission. Our ongoing working is focused on preparing amphiphilic copolymers and corresponding dual responsive micelles in aqueous system.
AcknowledgmentThe authors are grateful for financial support by the National Science Foundation of China (No. 21604071).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi: https://doi.org/10.1016/j.cclet.2019.03.053.
| [1] |
F.D. Jochum, P. Theato, Chem. Soc. Rev. 42 (2013) 7468-7483. DOI:10.1039/C2CS35191A |
| [2] |
J. Li, C. Nagamani, J.S. Moore, Acc. Chem. Res. 48 (2015) 2181-2190. DOI:10.1021/acs.accounts.5b00184 |
| [3] |
M.M. Russew, S. Hecht, Adv. Mater. 22 (2010) 3348-3360. DOI:10.1002/adma.200904102 |
| [4] |
F. Li, C. Xie, Z. Cheng, H. Xia, Ultrason. Sonochem. 30 (2016) 9-17. DOI:10.1016/j.ultsonch.2015.11.023 |
| [5] |
M.A. Ayer, Y.C. Simon, C. Weder, Macromolecules 49 (2016) 2917-2927. DOI:10.1021/acs.macromol.6b00418 |
| [6] |
M.J. Robb, J.S. Moore, J. Am. Chem. Soc. 137 (2015) 10946-10949. DOI:10.1021/jacs.5b07345 |
| [7] |
A.B. Seabra, N. Durán, J. Mater. Chem. 20 (2010) 1624-1637. DOI:10.1039/B912493B |
| [8] |
Y. Deng, F. Jia, S. Chen, et al., Biomaterials 187 (2018) 55-65. DOI:10.1016/j.biomaterials.2018.09.043 |
| [9] |
F. Ji, Y. Deng, J. Ji, Mater. Chem. Front. 2 (2018) 830-834. DOI:10.1039/C8QM00013A |
| [10] |
B.E. Mann, Organometallics 31 (2012) 5728-5735. DOI:10.1021/om300364a |
| [11] |
F. Zobi, Future Med. Chem. 5 (2013) 175-188. DOI:10.4155/fmc.12.196 |
| [12] |
N.E. Brückmann, M. Wahl, G.J. Reiß, et al., Eur. J. Inorg. Chem. 2011 (2011) 4571-4577. |
| [13] |
R. Sakla, D.A. Jose, ACS Appl. Mater. Inter. 10 (2018) 14214-14220. DOI:10.1021/acsami.8b03310 |
| [14] |
N. Zhou, L. Peng, S. Salgado, J. Yuan, X. Wang, Angew. Chem. Int. Ed. 56 (2017) 6246-6250. DOI:10.1002/anie.201611486 |
| [15] |
Y. Gong, Y. Zhang, W. Xiong, et al., J. Am. Chem. Soc. 139 (2017) 10649-10652. DOI:10.1021/jacs.7b06261 |
| [16] |
K. Matyjaszewski, J. Xia, Chem. Rev. 101 (2001) 90-146. |
| [17] |
M. Karman, E. Verde-Sesto, C. Weder, ACS Macro Lett. 7 (2018) 1028-1103. DOI:10.1021/acsmacrolett.8b00520 |
| [18] |
R. Tong, X. Lu, H. Xia, Chem. Comm. 50 (2014) 3575-3578. DOI:10.1039/c4cc00103f |
| [19] |
S.L. Potisek, D.A. Davis, N.R. Sottos, S.R. White, J.S. Moore, J. Am. Chem. Soc. 129 (2007) 13808-13809. DOI:10.1021/ja076189x |
| [20] |
A. Poloukhtine, V.V. Popik, J. Org. Chem. 20 (2003) 7833-7840. |
| [21] |
D.C. Church, G.I. Peterson, A.J. Boydston, ACS Macro Lett. 3 (2014) 648-651. DOI:10.1021/mz5003068 |
| [22] |
U. Hasegawa, A.J. van der Vlies, E. Simeoni, C. Wandrey, J.A. Hubbell, J. Am. Chem. Soc. 132 (2010) 18273-18280. DOI:10.1021/ja1075025 |
| [23] |
G. Dordelmann, H. Pfeiffer, A. Birkner, U. Schatzschneider, Inorg. Chem. 50 (2011) 4362-4367. DOI:10.1021/ic1024197 |
| [24] |
H.Y. Duan, Y.X. Wang, L.J. Wang, et al., Macromolecules 50 (2017) 1353-1361. DOI:10.1021/acs.macromol.6b02370 |
| [25] |
P.A.R. Glynn, B.M.E. Van Der Hoff, P.M. Reilly, J. Macromol. Sci. A 6 (1972) 1653-1664. DOI:10.1080/10601327208056918 |
| [26] |
S.W. Staley, T.D. Norden, W.H. Taylor, M.D. Harmony, J. Am. Chem. Soc. 109 (1987) 7641-7647. DOI:10.1021/ja00259a011 |