 2020, Vol. 31
2020, Vol. 31 


b CAS Key Lab of Low-Carbon Conversion Science and Engineering, Shanghai Advanced Research Institute, Chinese Academy of Sciences, Shanghai 201210, China;
c School of Physical Science and Technology, ShanghaiTech University, Shanghai 201210, China
With the technological maturation of shale gas recovery and the global demand to low-carbon development, CH4 (major component of natural gas and shale gas) occurred to be a promising resource in energy and chemical industries. It was estimated that the production and consumption of CH4 will increase drastically in the following decades [1]. However, CH4 has a higher greenhouse effect than CO2(25 times), therefore, a significant amount of low-grade CH4 was flared at the production site [2]. This is simply because the state-of-the-art technologies for CH4conversion rely on highly-centralized process with very large scale, and long-distance transportation of CH4 in gas phase is either cost-ineffective or impractical in some cases. Consequently, alternative approaches for CH4 conversion is highly desired [3].
Direct methane to methanol (DMTM) was proposed as a "dreaming" process for methane utilization. This process converts CH4 directly to liquid methanol without the production of syngas as an intermediate. Because of the abundant options of downstream methanol conversion, the DMTM process was regarded as "a paradigm shift in petrochemical technology" [4].
Unfortunately, activation of CH4 is not straightforward due to its inertness [5], and DMTM is particularly difficult because the target product, CH3OH, is more prone to oxidation than CH4. Consequently, the process needs to activate the C–H bond on one hand, and avoid over-oxidation of CH3OH on the other [4]. In nature, DMTM can be successfully performed by an enzymatic system called methane monooxygenase (MMO). The active sites in these systems were identified as diiron or dicopper centers with oxygen bridges [6, 7]. Inspired by these structures, ion and copper exchanged zeolites were extensively investigated as nature-mimicking materials to perform DMTM in a stepwise manner [8-25]. Among the proposed metal and zeolite combinations, Cu exchanged mordenite (MOR) showed outstanding performance [10, 11, 18, 20, 23, 24]. Upon activation (normally in an oxidative atmosphere at elevated temperatures), centers such as [Cu2(μ-O)2]2+, [Cu2(μ-O)]2+, [Cu3(μ-O)3]2+, or even CuOx clusters can be generated, which are responsible to activate the C–H bond in the subsequent CH4 reaction step [5, 8-15, 26-29].
Previous reports have established solid conclusion that speciation of the Cu sites is of great importance in determining their DMTM performance. In this context, the Cu exchange process can considerably affect the loading of metal and thus formation of different Cu sites. This has been well-demonstrated by: (1) using MOR zeolites with different Si/Al ratios [11, 18, 24]; (2) conduct the ion exchange at different pH or even in solid phase [15]. Different Cu salts were also used in previous publications, however, comparison of the effect of different anions under otherwise identical conditions was rarely reported. Herein, Cu@MOR catalysts were prepared by ion exchange of MOR zeolite with different Cu salts, while all the other preparation conditions were kept the same. The resulted samples were characterized and their performance in DMTM was investigated.
The Cu@MOR catalysts were prepared by ion-exchange of commercialized MOR zeolite with different Cu salts. The obtained samples were designated as Cu@MOR-A, Cu@MOR-C, Cu@MOR-N, and Cu@MOR-S when Cu(CH3COO)2, CuCl2, Cu(NO3)2 and CuSO4 were used, respectively. Detailed preparation methods can be found in Supporting information.
Fig. 1a shows the XRD patterns of the parent MOR zeolite and Cu-exchanged samples. All the diffraction peaks can be indexed exclusively to orthorhombic mordenite. No peak corresponding to Cu-related species, such as face centered cubic Cu, body centered cubic Cu2O, or monoclinic CuO was visible, indicating their absence in form of large particles (> 3 nm) [10, 11]. Porosity of the sample was verified by N2 physisorption at -196 ℃. As can be found from Fig. 1b, Type I isotherms were obtained for all of the samples, indicating their microporous nature. The parent MOR zeolite has a pore volume of 0.248 cm3/g, and loading of Cu did not cause any significant compromise on porosity. Fig. S1 (Supporting information) and Fig. 2 present the SEM images of the samples. The MOR zeolite exhibited morphology of fused crystallites in μm-scale. After introduction of Cu, no morphological change can be observed, evidenced again that the MOR framework remained intact, and any heterogeneity upon ion exchange can be ruled out. According to ICP-OES analysis, the Cu loading achieved 3.36, 1.77, 1.58, and 1.24 wt% for Cu@MOR-A, Cu@MOR-C, Cu@MOR-N, and Cu@MOR-S, respectively. Note that the DMTM catalytic performance does not depend only on Cu loading.

|
Download:
|
| Fig. 1. Characterization of the samples. (a) XRD patterns and (b) N2 adsorption isotherms (-196 ℃). The isotherms of Cu@MOR-A, Cu@MOR-C, Cu@MOR-N and Cu@MOR-S are vertically shifted by 140, 280, 420 and 560 cm3/g, respectively. | |
{kind=link}

|
Download:
|
| Fig. 2. SEM images of (a) Cu@MOR-A, (b) Cu@MOR-C, (c) Cu@MOR-N and (d) Cu@MOR-S. | |
{kind=link}
The catalytic performance of the samples towards DMTM was investigated on a fixed bed reactor with the widely used cyclic manner. Every cycle includes four steps (Fig. S2 in Supporting information) [8]: (1) Activation of the catalyst in O2 flow (100 mL/min) for 2 h at elevated temperature (either 400, 450, or 500 ℃); (2) Purge with He (100 mL/min) and cooling down to 200 ℃; (3) Feed 50 mL/min of pure CH4 at 200 ℃ for 0.5 h; (4) CH3OH desorption by purging with H2O saturated He (100 mL/min) at 200 ℃ for 0.5 h, and then increase the temperature to 400 ℃ (5 ℃/min) and hold for 2 h. The produced CH3OH was collected in a cold trap and analyzed by gas chromatography.
Firstly, capability of the Cu@MOR in catalyzing DMTM was demonstrated. All of the catalysts were activated in pure O2 at 400 ℃, and then submitted to the following DMTM steps. At such conditions, CH3OH yields achieved 26, 76, 29 and 48 μmol/g cat for Cu@MOR-A, Cu@MOR-C, Cu@MOR-S, and Cu@MOR-N, respectively. These values are comparable with previous publication [9]. Production of CH3OH during steam purging was also monitored by on-line MS. In Fig. S3 (Supporting information), the m/z = 31 signal intensified after the introduction of steam to the catalyst bed, indicating the desorption of CH3OH from the catalyst, which continued with the increasing of temperature. At ca. 320–340 ℃, a small amount of CO2 could be detected due to the unavoidable over-oxidation of some intermediates. Overall, the CH3OH selectivity was estimated to be 85%–90% in all the DMTM cycles reported in this work, the only by-product is CO2, no dimethyl ether was detected by GC analysis [30].
For the stepwise DMTM process, catalyst activation is one of the determining steps to dictate the speciation of Cu sites, which in turn played a pivotal role in affecting the CH3OH yield [9-11]. Therefore, the optimum activation temperatures for each of the Cu@MOR catalysts should be established. To this end, three consecutive DMTM cycles were carried out by using 400, 450, and 500 ℃ as the activation temperatures over each of the catalyst. From Fig. 3a, it can be found that increasing the activation temperature from 400 ℃ to 450 ℃ led to larger CH3OH yields, up to 95 μmol/g for Cu@MOR-C. This observation is in agreement with literatures, which can be related to the formation of larger number of active Cu sites at elevated temperatures [24]. Upon further increase of activation temperature to 500 ℃, decrease of CH3OH production was obtained except Cu@MOR-A. Consequently, it can be concluded that an appropriately high temperature for activation of the Cu@MOR catalysts, 450 ℃ in our cases, is necessary to achieve high DMTM performance.

|
Download:
|
| Fig. 3. Catalytic performance of the Cu@MOR catalysts. (a) Effect of activation temperature. (b) Effect of Cu exchange times. | |
{kind=link}
After the three consecutive DMTM cycles under different activation temperatures were finished, one additional cycle with 450 ℃ activation was carried out. As compared with the first cycle with 450 ℃ activation, very similar CH3OH production was obtained for Cu@MOR-C, Cu@MOR-N, and Cu@MOR-S (Fig. 3a, open symbols). This result indicates the formation and consumption of Cu active sites during DMTM process is reversible. It is highly possible that the "less active" sites formed at 500 ℃ activation was "used up" during the CH4 contact procedure, and the Cu species went back to a state that can be re-converted again to more active form by 450 ℃ activation. According to Alayon et al. work [28], we attribute the formation and consumption of active Cu sites to the reversible redox interconversion of Cu(Ⅱ) and Cu(Ⅰ).
Very interestingly, when Cu@MOR-A was re-activated at 450 ℃, significantly higher CH3OH production as compared to the 1st 450 ℃ activated cycle was obtained, achieving a promisingly high value of 131 μmol/g, which is among the highest in the open literatures [18, 25]. Gradual enhancement of CH3OH production during DMTM cycles was previously reported by several authors [8-11]. This observation can be related to the dynamic speciation of Cu active sites during the repeated DMTM cycles. For example, Xamena et al. and Palomino et al. reported that oxidation of Cu loaded zeolite by O2 can be facilitated by previous hydration [31, 32]. This is to say that during the DMTM steps, CH3OH extraction by steam purging may benefit the oxidative activation of the Cu sites in the subsequent cycle, leading to more active catalytic centers and thus higher CH3OH production. The above assumption was experimentally demonstrated by Bozbag and co-workers, their XANES results identified roughly 30% increase of the active Cu sites in a second DMTM cycle as compared with the first one [11]. We attribute the observed increase of CH3OH production over Cu@MOR-A at identical conditions to similar reasons, namely during the DMTM cycles, re-distribution and/or re-speciation of Cu gradually led to the formation of species that can be more readily oxidized to highly active configurations.
Apart from the activation condition discussed above, Cu loading also played a decisive role to the DMTM performance of the Cu@MOR catalysts. As such, three ion exchange procedures were carried out by using different Cu salts to afford another four samples: Cu@MOR-A3, Cu@MOR-C3, Cu@MOR-N3 and Cu@MOR-S3. According to the ICP-OES measurements, loading of Cu was effectively increased to 4.50, 4.52, 3.95 and 3.58 wt% for Cu@MOR-A, Cu@MOR-C, Cu@MOR-N, and Cu@MOR-S, respectively. The XRD patterns (Fig. S4 in Supporting information) excluded formation of any large CuOx domains. DMTM reaction was performed with an activation temperature of 450 ℃, and the results were showed in Fig. 3b for clearer comparison. Surprisingly, CH3OH production over Cu@MOR-C3 and Cu@MOR-N3 decreased as compared with the corresponding samples with only one ion exchange process, probably due to pore blockage and/or formation of metal clusters too small to be detected by XRD. On the other hand, Cu@MOR-A3 showed higher performance with a CH3OH production of 101 μmol/g, higher than the first 450 ℃ activated cycle for Cu@MOR-A.
The above results indicated that when Cu(CH3COO)2 was used as the Cu precursor, high methanol yields can be obtained, and evolution of the original Cu species to more active structure can be achieved. Therefore, we came to the conclusion that Cu(CH3COO)2 occurs to be the best precursor for the preparation of Cu@MOR catalyst for DMTM. It has been demonstrated that under atmospheric pressure, the MeOH yield during DMTM over the Cu exchanged MOR correlated closely to the oxygen bonded Cu2+ clusters, such as [Cu2(μ-O)2]2+, [Cu2(μ-O)]2+, and [Cu3(μ-O)3]2+ [5, 8-15, 26-29]. Therefore, the high performance of the Cu(CH3COO)2 derived catalysts can be ascribed to more favorably formation of such structures. This effect may be attributed to higher affinity of CH3COO- towards protons: Firstly, since the CH3COO- ion has a higher tendency to associate with H+ as compared with Cl-, NO3-, and SO42-, more exchangeable protons in the MOR framework can be extracted, leading to higher flexibility for the loading of Cu2+; Secondly, combination of CH3COO- and H+ in the solution phase increased the concentration of OH-, and thus facilitated the formation of [Cu(OH)]+, which was reported to be a more efficient intermediate for the formation of the Cu sites with higher DMTM activity [18].
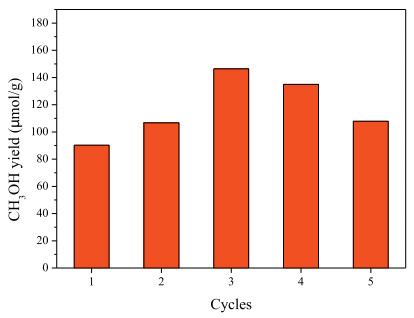
Last but not the least, the cycling stability of Cu@MOR-A3 was investigated. Five DMTM cycles were performed, it can be seen from Fig. 4 that very high and stable CH3OH production of 117 ± 28 μmol/g was maintained during the cycles. The sample after stability test was characterized by XRD and SEM (Fig. 5), no degradation on phase structure and morphology was noticed. These results demonstrated the great promise of the Cu@MOR-A3 for further DMTM studies.

|
Download:
|
| Fig. 4. DMTM stability of Cu@MOR-A3. | |
{kind=link}

|
Download:
|
| Fig. 5. Characterization of Cu@MOR-A3 after 5 DMTM cycles. (a) XRD patterns, and (b) SEM image. | |
{kind=link}
In summary, we prepared a series of Cu@MOR catalysts from different Cu salts by ion exchange with MOR zeolite, and the effect of counter ions on the catalytic performance in direct conversion of CH4 to CH3OH was investigated. Results from characterization and reaction evaluation indicated that the speciation of Cu centers was influenced by the used counter ions. This originates probably from (1) different capability in extraction of protons from MOR, and (2) varied hydrolysis state of the Cu2+ in aqueous solution. Finally, Cu@MOR-A3 prepared from Cu(CH3COO)2 was showed to have stable CH3OH yields of 117 ± 28 μmol/g in 5 consecutive cycles, among the highest in open publications.
AcknowledgementThis paper is sponsored by Chinese Academy of Sciences Youth Innovation Promotion Association Funding (No. 2018329).
Appendix A. Supplementary dataSupplementary material related to this article can befound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.03.039.
| [1] |
U.S. Energy Information Administration, International Energy Outlook (2018). |
| [2] |
C.D. Elvidge, M. Zhizhin, K. Baugh, et al., Energies 9 (2016) 14. |
| [3] |
J.R. Rostrup-Nielsen, J. Sehested, J.K. Nørskov, Adv. Catal. 47 (2002) 65-139. |
| [4] |
M. Ravi, M. Ranocchiari, J.A. van Bokhoven, Angew. Chem. Int. Ed. 56 (2017) 16464-16483. DOI:10.1002/anie.201702550 |
| [5] |
P. Tomkins, M. Ranocchiari, J.A. Van Bokhoven, Acc. Chem. Res. 56 (2017) 418-425. DOI:10.1021/acs.accounts.6b00534 |
| [6] |
A.C. Rosenzweig, C.A. Frederick, S.J. Lippard, P. Nordlund, Nature 366 (1993) 537-543. DOI:10.1038/366537a0 |
| [7] |
S. Friedle, E. Reisner, S.J. Lippard, Chem. Soc. Rev. 39 (2010) 2768-2779. DOI:10.1039/c003079c |
| [8] |
V.L. Sushkevich, D. Palagin, M. Ranocchiari, et al., Science 356 (2017) 523-527. DOI:10.1126/science.aam9035 |
| [9] |
P. Tomkins, A. Mansouri, S.E. Bozbag, et al., Angew. Chem. Int. Ed. 55 (2016) 5467-5471. DOI:10.1002/anie.201511065 |
| [10] |
E.M.C. Alayon, M. Nachtegaal, M. Ranocchiari, et al., Chem. Commun. 48 (2012) 404-406. DOI:10.1039/C1CC15840F |
| [11] |
S.E. Bozbag, E.M.C. Alayon, J. Pechácek, et al., Catal. Sci. Technol. 6 (2016) 50119-5022. DOI:10.1039/C6CY00041J |
| [12] |
M.B. Park, S.H. Ahn, A. Mansouri, et al., ChemCatChem 9 (2017) 3705-3713. DOI:10.1002/cctc.201700768 |
| [13] |
E.M.C. Alayon, M. Nachtegaal, A. Bodi, et al., Phys. Chem. Chem. Phys. 17 (2015) 7681-7693. DOI:10.1039/C4CP03226H |
| [14] |
M.A.C. Markovits, A. Jentys, M. Tromp, et al., Top. Catal. 59 (2016) 1554-1563. DOI:10.1007/s11244-016-0676-x |
| [15] |
T. Sheppard, C.D. Hamill, A. Goguet, et al., Chem. Commun. 50 (2014) 11053-11055. DOI:10.1039/C4CC02832E |
| [16] |
M.J. Wulfers, S. Teketel, B. Ipek, et al., Chem. Commun. 51 (2015) 4447-4450. DOI:10.1039/C4CC09645B |
| [17] |
B. Ipek, R.F. Lobo, Chem. Commun. 52 (2016) 13401-13404. DOI:10.1039/C6CC07893A |
| [18] |
S. Grundner, M.A.C. Markovits, G. Li, et al., Nat. Commun. 6 (2015) 7546. DOI:10.1038/ncomms8546 |
| [19] |
K. Narsimhan, K. Iyoki, K. Dinh, et al., ACS Cent. Sci. 2 (2016) 424-429. DOI:10.1021/acscentsci.6b00139 |
| [20] |
S. Grundner, W. Luo, M. Sanchez-Sanchez, et al., Chem. Commun. 52 (2016) 2553-2556. DOI:10.1039/C5CC08371K |
| [21] |
D.K. Pappas, E. Borfecchia, M. Dyballa, et al., J. Am. Chem. Soc. 139 (2017) 14961-14975. DOI:10.1021/jacs.7b06472 |
| [22] |
T. Sheppard, H. Daly, A. Goguet, et al., ChemCatChem 8 (2016) 562-570. DOI:10.1002/cctc.201500980 |
| [23] |
Y. Kim, T.Y. Kim, H. Lee, et al., Chem. Commun. 53 (2017) 4116-4119. DOI:10.1039/C7CC00467B |
| [24] |
H.V. Le, S. Parishan, A. Sagaltchik, et al., ACS Catal. 7 (2017) 1403-1412. DOI:10.1021/acscatal.6b02372 |
| [25] |
D.K. Pappas, A. Martini, M. Dyballa, et al., J. Am. Chem. Soc. 140 (2018) 15270-15278. DOI:10.1021/jacs.8b08071 |
| [26] |
D. Palagin, A.J. Knorpp, A.B. Pinar, et al., Nanoscale 9 (2017) 1144-1153. DOI:10.1039/C6NR07723D |
| [27] |
S.E. Bozbag, P. Šot, M. Nachtegaal, et al., ACS Catal. 8 (2018) 5721-5731. DOI:10.1021/acscatal.8b01021 |
| [28] |
E.M.C. Alayon, M. Nachtegaal, A. Bodi, et al., ACS Catal. 4 (2013) 16-22. DOI:10.1021/cs400713c |
| [29] |
D. Palagin, A.J. Knorpp, A.B. Pinar, et al., Nanoscale 9 (2017) 1144-1153. DOI:10.1039/C6NR07723D |
| [30] |
E.V. Starokon, M.V. Parfenov, L.V. Pirutko, et al., J. Phys. Chem. C 115 (2011) 2155-2161. DOI:10.1021/jp109906j |
| [31] |
F.X. Llabrés i Xamena, P. Fisicaro, G. Berlier, et al., J. Phys. Chem. B 107 (2003) 7036-7044. DOI:10.1021/jp0275847 |
| [32] |
G.T. Palomino, P. Fisicaro, S. Bordiga, et al., J. Phys. Chem. B 104 (2000) 4064-4073. DOI:10.1021/jp993893u |