 2020, Vol. 31
2020, Vol. 31 

b International Center of Future Science, Jilin University, Changchun 130012, China
As a major component of greenhouse gases, carbon dioxide is a main cause of global warming [1-6]. Zeolitic imidazolate frameworks are promising porous materials for CO2 capture because of their large accessible volumes, tunable functional groups, and relatively high thermal and chemical stability [7-14]. In particular, the properties of ZIFs are essentially determined by their network topologies [15, 16]. To this end, many computational efforts have been made to build a number of hypothetical ZIF models from specific topologies, from which candidates with the most promising CO2 capture performance may be identified [17-20]. For instance, Smit et al. studied 1000 hypothetical ZIFs for CO2 capture and identified 50 of them with the smallest parasitic energy as candidate materials [21]; Calero et al. constructed 197 hypothetical ZIF models from known zeolites and identified 10 candidates with the best Henry's constant ratio or adsorption selectivity [19]. Le et al. built 18 hypothetical ZIF models from 3 network topologies and identified two of them as potential candidates for CO2 capture [20]. Despite all the success, the number of explored ZIF models is rather limited in comparison with other MOF materials [22, 23].
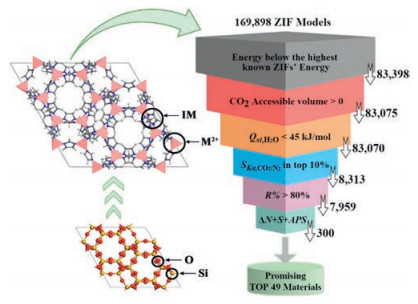
Here, we report a high-throughput study in model-building and screening of new ZIF structures for CO2 capture from flue gas. We built 169, 898 ZIF models from 84, 949 hypothetical zeolite networks. These ZIF models were further evaluated by lattice energies, accessible volume to CO2, the isosteric adsorption heat (Qst) of H2O, the ratio of Henry's constant (SKH) of CO2/N2, percent regenerability (R%), CO2 working capacity (ΔNCO2), CO2/N2 adsorption selectivity (SCO2/N2) and adsorbent performance score (APS). Among such a large number of ZIF models we explored, we were able to identify 49 hydrophobic ZIF structures that might be superior to experimentally realised ZIF structures built from the same imidazolate linkers for CO2 capture (Scheme 1).

|
Download:
|
| Scheme 1. Schematic diagram of high-throughput model-building and screening of hypothetical ZIF models. | |
{kind=link}
The hypothetical zeolite networks we began with were obtained in our previous study, including 84, 292 ABC-6 networks generated by structure enumeration [24] and 657 tetrahedral networks generated by automated atom assembly [25, 26]. We wrote a perl script to build 169, 898 ZIF models automatically from zeolite networks by replacing the vertex Si atoms and the bridging O atoms in zeolite networks by Zn or Cu cations and imidazolate linkers, respectively. The ZIF models derived from ABC-6 networks are named ABC-ZIFs hereafter and those from automated atom assembly are named AAA-ZIFs. Then, the unit cell dimensions of the original zeolite networks were expanded by 1.9 times to approximate those of ZIF structures. All ZIF models were further optimised using the UNIVERSAL force field [27]. The highest allowed symmetries were imposed to these models throughout geometry optimisation. More details are shown in Table S1 (Supporting information). On the other hand, we optimised 24 Zn-ZIF and 5 Cu-ZIF structures that were experimentally realised [22]. All these calculations were conducted using Materials Studio software package [28]. Considering the fact that the hypothetical ZIF models with high lattice energies would be unstable and difficult to realise in experiment, we set the upper limits for the lattice energies of hypothetical ZIFs as the highest lattice energies observed in realised ZIFs. By doing this, we narrowed down the list of stable ZIFs to 83, 398 (Fig. S1 in Supporting information).
We calculated the pore-limiting diameters (PLDs) and the largest-cavity diameters (LCDs) for all stable hypothetical ZIFs using program Zeo++ [29]. The results are shown in Fig. S2a (Supporting information). Because all our ZIF models were built from zeolite networks, most of them possess large void spaces. In comparison to AAA-ZIFs (PLD: 2.5-7.5 Å; LCD: 5-10 Å), ABC-ZIFs exhibit larger void space for guest species (PLD: 4-7 Å; LCD: 10.5-14 Å) because of their featured cage structures. Then, the CO2 accessible volumes and surface areas for these ZIFs were calculated using a spherical probe with a diameter of 3.3 Å, which corresponded to the kinetic diameter of CO2 (Fig. S2b in Supporting information). 323 ZIF models were removed because of the absence of pores accessible to CO2. We also calculated the CO2 accessible volumes for experimentally realised ZIFs. Half of the realised Zn-ZIFs (12/24) and most of the realised Cu-ZIFs (4/5) were removed because of the absence of accessible pores.
To evaluate the CO2 capture performance of these ZIF models, we conducted single-component Grand Canonical Monte Carlo (GCMC) simulations for the remaining 83, 075 ZIF models at 298 K. All simulations were performed using Materials Studio software package. All force field parameters were taken from literature [30-33]. Detailed information can be found in Table S2 (Supporting information). First, we calculated the isosteric adsorption heat, Qst, and the Henry's constant, KH, H2O of H2O molecules in these ZIF models at infinite dilution. The results are shown in Fig. S3 (Supporting information). Only five of the remaining hypothetical ZIF models exhibited isosteric adsorption heat greater than 45 kJ/mol, and over 80% of them less than 30 kJ/mol. As expected, most of our ZIF models were not hydrophilic, indicating their potential applications under real flue gas environment with certain humidity [34].
Then, we separately calculated the adsorption of CO2 and N2 molecules of the remaining 83, 070 ZIF models and obtained their Henry's constant, i.e., KH, CO2 and KH, N2 respectively. The ratio of Henry's constant of CO2/N2 was calculated using the formula:

|
(1) |
which represented the ideal selectivity of the adsorbent at infinite dilution. The ratio of Henry's constant would be a good approximation of the true separation capability of the adsorbent under practical conditions, and its computational cost is much lower than regular multi-component GCMC simulations [35, 36]. According to our calculations, the Henry's constant ratios of the 83, 070 ZIF structures ranged from 5.83 to 8525.37 (Fig. S4 in Supporting information). We kept the top 10% models with the highest SK for further simulations. Note that two of the realised ZIF structures were removed at this step because of the low Henry's constant ratios.
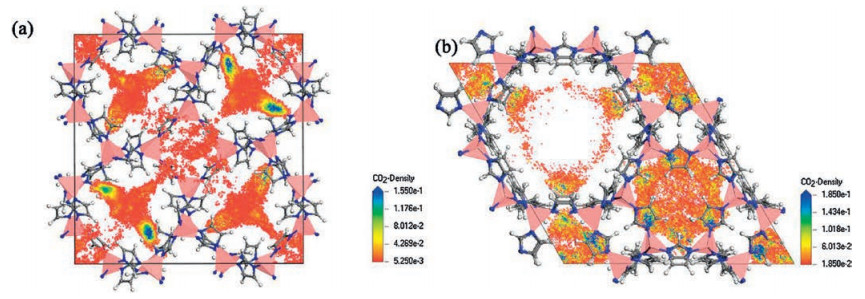
Next, we performed multi-component GCMC simulations for the remaining 8313 hypothetical ZIF models and 11 already-realised structures with the highest SKH in CO2/N2/H2O ternary systems. The simulations were conducted at 10 bar and 1 bar, corresponding to the gas adsorption pressure and the desorption pressure, respectively. The CO2:N2 molar ratio was set to 1:9, and a relative humidity of 80% was maintained in correspondence with the content of flue gas [37, 38]. Fig. 1 shows the density distribution of CO2 molecules in two selected ZIF models during GCMC simulations in CO2/N2/H2O ternary systems. The CO2 molecules are mainly located close to the imidazolate linkers. Such affinity can also be observed in the molecular dynamics trajectories of individual CO2 molecules (Fig. S5 in Supporting information).

|
Download:
|
| Fig. 1. Density distribution of the centre-of-mass of CO2 molecules along the c-axis in the unit cells of (a) zif_7322-36172 and (b) zif_001010101010101 at 298 K and 10 bar. Colour notation: Zn, pink tetrahedral; C, grey spheres; N, blue spheres; H, white spheres. | |
{kind=link}
Using the simulation date obtained from CO2/N2/H2O ternary systems, we calculated the percent regenerability (R%), which is defined as the ratio of the working capacity (ΔNCO2) and the adsorption capacity (NCO2ads) at high pressure:

|
(2) |

|
(3) |
where NCO2ads and NCO2des are loadings at adsorption and desorption conditions, respectively. R% represents the fraction of the adsorption sites that are regenerable at low pressure [39, 40]. The results are shown in Fig. S6 (Supporting information). 7959 of our remaining 8313 ZIF models exhibited R% ≥ 80%. In comparison, only 2 of the already-realised ZIF structures exhibited R% ≥ 80%. Because materials with low R% would not be suitable for practical cyclic utilization [39-41], we screened out 354 hypothetical models with R% < 80% and kept 7959 models for further evaluation. All the remaining high-R% ZIFs exhibited CO2 isosteric adsorption heat less than 35 kJ/mol (Fig. S6), so the adsorbed CO2 molecules could be removed easily at low pressure. This might be due to the highly porous nature of our ZIF models that were built from open-framework zeolite networks.
To identify the best structures for CO2 capture from the remaining ZIFs, we further calculated adsorption selectivity (SCO2/N2) and APS as follows [37, 39-42]:

|
(4) |

|
(5) |
where NCO2ads and NN2des are loadings at adsorption and desorption conditions, respectively; y is the mole fraction in the flue gas phase. All these data were calculated from GCMC simulations in CO2/N2/H2O ternary systems.
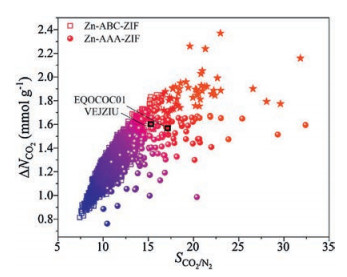
The results are shown in Fig. 2. The maximum adsorption working capacity ΔNCO2 of our hypothetical ZIF structures reached 2.37 mmol/g, which was much higher than that for experimentally realised structures (1.61 mmol/g). The highest adsorption selectivity SCO2/N2 for our hypothetical ZIF models was 31.83, and 17.15 for already-realised ZIFs. The best APS for hypothetical and experimentally realised ZIFs were 68.70 mmol/g and 27.03 mmol/g, respectively. In general, the performance of the remaining hypothetical ZIFs was significantly better than already-realised ones. By ranking their adsorption working capacity, adsorption selectivity, and adsorbent performance score, we identified 49 hypothetical ZIF models that were among the top 100 in all categories. Detailed information of the 49 structures are listed in Table S3 (Supporting information). The isosteric CO2 adsorption heat for the 49 hypothetical ZIF models were within the range of 24.26-30.99 kJ/mol, which were close to the ideal values reported by Snurr et al. [37].

|
Download:
|
| Fig. 2. Adsorption selectivity (SCO2/N2) versus working capacity (ΔNCO2) for 7959 hypothetical ZIFs and 2 already-realised ZIFs with R% ≥ 80%. The adsorbent performance score of all ZIFs are shown in different colours from blue to red. The top 49 materials were marked as stars. | |
{kind=link}
In general, AAA-ZIF models exhibited better CO2 capture performance than ABC-ZIFs, especially in adsorption selectivity and adsorbent performance score. This might be because the AAA-ZIF models possessed smaller void space than ABC-ZIFs, and relatively smaller pore openings are usually crucial for selective adsorption [4, 37]. Although our hypothetical ZIF models did not exhibit the best CO2 selectivity among all types of ZIF materials (Table S4 in Supporting information), the results we obtained here were quite acceptable because we did not include any additional functional groups in our ZIF models, which could dramatically increase the affinity to CO2 molecules [4, 9, 16, 43, 44].
Because of the lack of open metal site and amino group, the interactions between CO2 molecules and our ZIF models were mainly physical adsorptions. To further understand the contributions of van der Waals interactions and electrostatic interactions in adsorptions, we performed single-component GCMC simulations separately with and without charge assignment, and calculated the corresponding adsorption isotherms for two selected ZIF models with the best working capacity and selectivity at 298 K [44]. The results are shown in Fig. S7 (Supporting information). For N2 adsorption, electrostatic interactions almost had no contribution to the loading and isosteric heat for both ZIFs. For CO2 adsorption, van der Waals interactions still dominated. However, electrostatic interactions did influence CO2 adsorption capacity and isosteric heat, because CO2 possessed a higher quadrupolar moment than N2 [45, 46].
In conclusion, through high-throughput calculations, we constructed 169, 898 hypothetical ZIF models and identified the most promising candidates for CO2 capture from flue gas by step-wise screening. Our hypothetical ZIF models exhibited much better performance than already-realised ZIFs built from the same imidazolate linkers. Our approach enables efficient construction and evaluation of such a large number of ZIF models that have never been achieved before. This will accelerate the experimental discovery of novel ZIF materials with properties superior to existing ones. In this work, we have only considered imidazolate linkers. In future study, we will add functional groups with different affinities to CO2 to further tune the CO2 capture performance.
AcknowledgmentsThis work was supported by the National Key Research and Development Program of China (No. 2016YFB0701100), the National Natural Science Foundation of China (Nos. 21622102 and 21621001), the National 111 Project (No. B17020), Program for JLUSTIRT, and High Performance Computing Center of Jilin University.
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.04.025.
| [1] |
J. Li, Y. Ma, M.M. Colin, et al., Coord. Chem. Rev. 255 (2011) 1791-1823. DOI:10.1016/j.ccr.2011.02.012 |
| [2] |
O. Alduhaish, B. Li, H. Arman, et al., Chin. Chem. Lett. 28 (2017) 1653-1658. DOI:10.1016/j.cclet.2017.04.025 |
| [3] |
Q. Li, J. Duan, W. Jin, Chin. Chem. Lett. 29 (2018) 854-856. DOI:10.1016/j.cclet.2017.11.008 |
| [4] |
J. Yu, L. Xie, J. Li, et al., Chem. Rev. 117 (2017) 9674-9754. DOI:10.1021/acs.chemrev.6b00626 |
| [5] |
A. Modak, S. Jana, Microporous Mesoporous Mater. 276 (2019) 107-132. DOI:10.1016/j.micromeso.2018.09.018 |
| [6] |
W. Morris, N. He, K.G. Ray, et al., Chin. Chem. Lett. 29 (2018) 482-484. DOI:10.1016/j.cclet.2017.07.031 |
| [7] |
H. Hayashi, A.P. Cote, H. Furukawa, et al., Nat. Mater. 6 (2007) 501-506. DOI:10.1038/nmat1927 |
| [8] |
R. Banerjee, A. Phan, B. Wang, et al., Science 319 (2008) 939-943. DOI:10.1126/science.1152516 |
| [9] |
R. Banerjee, H. Furukawa, D. Britt, et al., J. Am. Chem. Soc. 131 (2009) 3875-3877. DOI:10.1021/ja809459e |
| [10] |
Y. Wang, L. Li, L. Yan, et al., Chin. Chem. Lett. 29 (2018) 849-853. DOI:10.1016/j.cclet.2017.09.057 |
| [11] |
A. Phan, C.J. Doonan, Uribe Romo F.J., et al., Acc. Chem. Res. 43 (2010) 58-67. DOI:10.1021/ar900116g |
| [12] |
N.T. Nguyen, H. Furukawa, F. Gandara, et al., Angew. Chem. Int. Ed. 53 (2014) 10645-10648. DOI:10.1002/anie.201403980 |
| [13] |
W. Lai, G. Zhuang, H. Tseng, M. Wey, J. Membr. Sci. 572 (2019) 410-418. DOI:10.1016/j.memsci.2018.11.010 |
| [14] |
W. Fan, Y. Wang, Z. Xiao, et al., Chin. Chem. Lett. 29 (2018) 865-868. DOI:10.1016/j.cclet.2017.11.020 |
| [15] |
C. Zheng, D. Liu, Q. Yang, et al., Ind. Eng. Chem. Res. 48 (2009) 10479-10484. DOI:10.1021/ie901000x |
| [16] |
M. William, H. Ning, G R.K., et al., J. Phys. Chem. C 116 (2012) 24084-24090. DOI:10.1021/jp307170a |
| [17] |
D.W. Lewis, A.R. Ruiz Salvador, A. Gómez, et al., CrystEngComm 11 (2009) 2272-2276. DOI:10.1039/b912997a |
| [18] |
L.M. Rodriguez Albelo, A.R. Ruiz Salvador, A. Sampieri, et al., J. Am. Chem. Soc. 131 (2009) 16078-16087. DOI:10.1021/ja905009e |
| [19] |
A.P. Gomez, S. Hamad, M. Haranczyk, et al., Dalton Trans. 45 (2016) 216-225. DOI:10.1039/C5DT04012D |
| [20] |
H.T. Hoang, H.L. Nguyen, T.B. Phan, et al., J. Phys. Chem. C 122 (2018) 23543-23553. DOI:10.1021/acs.jpcc.8b07159 |
| [21] |
L.C. Lin, A.H. Berger, R.L. Martin, et al., Nat. Prod. Sci. 11 (2012) 633-641. |
| [22] |
F.H. Allen, Acta Crystallogr. Sect. B: Struct. Sci. B 58 (2002) 380-388. DOI:10.1107/S0108768102003890 |
| [23] |
C.E. Wilmer, M. Leaf, C.Y. Lee, et al., Nat. Chem. 4 (2011) 83-89. |
| [24] |
Y. Li, X. Li, J. Liu, et al., Nat. Commun. 6 (2015) 8328. DOI:10.1038/ncomms9328 |
| [25] |
Y. Li, J. Yu, R. Xu, J. Appl. Crystallogr. 45 (2012) 855-861. DOI:10.1107/S002188981201878X |
| [26] |
Y. Li, J. Yu, https://figshare.com/collections/Hypothetical_Zeolite_Structures/4424417, (2019 March).
|
| [27] |
A.K. Rappe, C.J. Casewit, K.S. Colwell, et al., J. Am. Chem. Soc. 114 (1992) 10024-10035. DOI:10.1021/ja00051a040 |
| [28] |
Studio Materials, Systèmes Dassault, San Diego, CA (2017). |
| [29] |
T.F. Willems, C.H. Rycroft, M. Kazi, et al., Microporous Mesoporous Mater. 149 (2012) 134-141. DOI:10.1016/j.micromeso.2011.08.020 |
| [30] |
S.L. Mayo, B.D. Olafson, W.A. Goddard, J. Phys. Chem. 94 (1990) 8897-8909. DOI:10.1021/j100389a010 |
| [31] |
J.G. Harris, K.H. Yung, J. Phys. Chem. 99 (1995) 12021-12024. DOI:10.1021/j100031a034 |
| [32] |
J. Pérez-Pellitero, H. Amrouche, F.R. Siperstein, et al., Chem. -Eur. J. 16 (2010) 1560-1571. DOI:10.1002/chem.200902144 |
| [33] |
W.L. Jorgensen, J. Chandrasekhar, J.D. Madura, et al., J. Chem. Phys. 79 (1983) 926-935. DOI:10.1063/1.445869 |
| [34] |
L. Ding, A.O. Yazaydin, Phys. Chem. Chem. Phys. 15 (2013) 11856-11861. DOI:10.1039/c3cp51003d |
| [35] |
E. Haldoupis, S. Nair, D.S. Sholl, J. Am. Chem. Soc. 134 (2012) 4313-4323. DOI:10.1021/ja2108239 |
| [36] |
S. Yang, X. Lin, W. Lewis, et al., Nat. Mater. 11 (2012) 710-716. DOI:10.1038/nmat3343 |
| [37] |
C.E. Wilmer, O.K. Farha, Y.S. Bae, et al., Energy Environ. Sci. 5 (2012) 9849-9856. DOI:10.1039/c2ee23201d |
| [38] |
S. Li, Y.G. Chung, R.Q. Snurr, Langmuir 32 (2016) 10368-10376. DOI:10.1021/acs.langmuir.6b02803 |
| [39] |
Y.S. Bae, R.Q. Snurr, Angew. Chem. Int. Ed. 50 (2011) 11586-11596. DOI:10.1002/anie.201101891 |
| [40] |
Z. Sumer, S. Keskin, Ind. Eng. Chem. Res. 55 (2016) 10404-10419. DOI:10.1021/acs.iecr.6b02585 |
| [41] |
C. Altintas, G. Avci, H. Daglar, et al., ACS Appl. Mater. Interfaces 10 (2018) 17257-17268. DOI:10.1021/acsami.8b04600 |
| [42] |
Z. Qiao, K. Zhang, J. Jiang, J. Mater. Chem. A 4 (2016) 2105-2114. DOI:10.1039/C5TA08984K |
| [43] |
H. Amrouche, S. Aguado, J. Pérez Pellitero, et al., J. Phys. Chem. C 115 (2011) 16425-16432. DOI:10.1021/jp202804g |
| [44] |
Y. Liu, J. Liu, Y.S. Lin, M. Chang, J. Phys. Chem. C 118 (2014) 6744-6751. DOI:10.1021/jp4113969 |
| [45] |
K.G. Ray, D. Olmsted, N. He, et al., Phys. Rev. B: Condens. Matter. 85 (2012) 085410. DOI:10.1103/PhysRevB.85.085410 |
| [46] |
R. Babarao, J. Jiang, J. Am. Chem. Soc. 131 (2009) 11417-11425. DOI:10.1021/ja901061j |