 2020, Vol. 31
2020, Vol. 31 

b College of Sciences & Institute for Sustainable Energy, Shanghai University, Shanghai 200444, China;
c Shanghai CENAT New Energy Co., Ltd., Shanghai 201815, China
In the electronic-driven society, new energy storage systems and technologies become more important with the growing demand for energy [1-3]. The achieving of excellent performance for energy storage devices is a burning issue. Energy storage and conversion systems such as batteries [4], fuel cells [5] and supercapacitors [6, 7] have shown the unique potential and various advantages in perspectives on the current challenges [8]. Energy storage/conversion systems, including Li-ion batteries, Li-sulfur batteries [9, 10], aqueous rechargeable Sodium-based batteries [11] and K-ion batteries [12] have been developed rapidly to satisfy global energy demands of electric automobiles. Lithium-ion batteries (LIBs) become an indispensable part of people's production and life, which have been widely used energy storage because of their low cost, long cycle life, high energy density and good reversibility [13]. Synthesis of novel and advanced rechargeable Li-ion batteries materials is imminent. Therefore, looking for such anode materials with high energy density, enhanced rate capabilities and superior safety features is needed [14]. Transition-metal oxides, such asCo3O4 [15] and MnO2 [16] are used as negative electrodes for LIBs with high theoretical capacitance because of their multiple oxidation states. Binary transition-metal oxides materials typically exhibit electrochemical Faradaic reactions and a higher electrical conductivity, which arises from the combined contributions from both metal atoms with rich chemical valence states [17].
Molybdenum-based oxides with superior performances as an anode material for LIBs/SIBs [18] attracted the attention of scientists. Due to the variable valency and diverse structures, composite materials can be obtained under different synthesis conditions [19]. Recently, binary metal oxides such as NiMoO4 [20], ZnMoO4 [21], MnMoO4 [22], Co-Se [23] and their composites have been reported owing to their higher capacity and the rich oxidation states to get superior electrochemical performance. Besides, unique nanostructured topographies have been developed to address issues related to the significant volume changes, the short diffusion length and larger reaction surface area [17]. Therefore, synthesizing nanostructured binary metal oxides with high electrochemical performance remains a challenge.
The inorganic crystal structure database (ICSD) [24] contains various metal oxides. Diverse molybdenum-based oxide material has been employed to improve the electrochemical properties of cathode material. Among them, tin oxides and molybdenum oxides with two-dimensional layered structure are electroactive materials with multiple oxidation states, which increases the electrode conductivity [18]. The vander Waals gaps between octahedron Mo–O sheets and Sn layers result in enhanced electrochemical property [25]. Moreover, different Tin molybdenum oxide nanostructures maybe result in different electrochemical behaviors.
In this work, we synthesized flake-like β-SnMoO4 via a facile one-step synthetic method by solid-state reactions (SSR) method. Here, we demonstrate the material as a novel anode material, and further study its crystalline structure and electrochemical properties. From a survey of the literature, it is the first report on the β-SnMoO4 composite as an anode material for LIBs. The present study aims to explore novel materials within Mo-based complex oxides. Through the control of these properties, it is possible providing one of the best routes for the design of new electrode materials for high-performance properties in future.
Commercial analytical grade reagents (AR) chemicals in our present work were used without further purification.
X-ray powder diffraction (XRD, Bruker D8 Discover) was used to examine the crystallographic structure of the product's diffractometer measured with Cu–Kα radiation (λ =1.54178 Å) in the 2θ range of 5°–90°. Raman spectra were recorded on a Jobin-Yvon Labram-HR confocal laser micro Raman spectrometer with an argon-ion laser at an excitation wavelength of 514.5 nm and a resolution factor of 1 cm−1 and were obtained in the range of 100–1500 cm−1. IR spectra were recorded on a Bruker Tensor 27 apparatus with MCT detector (64 scans, 4 cm−1). TG analysis was performed by instruments TG/DTA-7300. SEM and EDS spectrum were obtained by using a Hitachi S-3000 H scanning electron microscope (SEM Tech Solutions) and a Horiba EMAX X-ACT, respectively. TEM images were examined on a Shimadzu JEM-1200 EX with an accelerating voltage of 100 kV. XPS results were carried out using a Thermo ESCALAB 250 instrument.
The electrode was prepared from a combination of active composite materials with super P carbon and PVDF binder (7:2:1 wt%). The PVDF was dissolved in NMP, and a mixture of active material with super P carbon was added to the solution to produce a homogenous slurry. The slurry was coated on a Cu foil (current collector), dried under vacuum at 60 ℃ for 12 h. The mass loading of active material was ~1.0 mg/cm2. Before the discharge/charge process, the cell was aged for 12 h to ensure full absorption of the electrolyte into electrodes.
The electrochemical performance was evaluated via CR2025-type coin cell on a LAND battery testing system (CT2001A, China). Galvanostatic discharge/charge tests were performed in the voltage range of 0.01–2.50 V under constant current mode. Cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) tests were carried out on a CHI 760D electrochemical workstation at the cell voltage of 2.2 V in the frequency range from 100 kHz to 0.01 Hz. The mass of SnMoO4 material is about 0.6 mg. All of the electrochemical measurements were carried out at room temperature.
All the related DFT calculations as described are performed using the CASTEP [26, 27] (Cambridge Serial Total Energy Package) module in Material Studio 7.0. The crystal structure is obtained by modification and optimization from β-SnWO4 crystal. After changing W composition into Mo, the crystal cell is optimized using the generalized gradient approximation (GGA) with the Perdew-Bruke-Ernzerof (PBE) exchange correlation function [28], based on the ultra-soft pseudopotential method. The double numeric polarization (DNP) basis set [29] was used for describing atomic orbitals. A real-space orbital global cutoff of 4.5 Å was applied. The convergence thresholds for optimization were 10−5 (energy), 2×10−3 (gradient), and 5×10−3 (displacement). The XRD was calculated for the optimized cell with the Reflex module. The powder refinement was performed using Rietveld type.
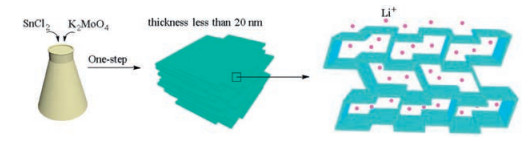
The overall synthetic strategy for the Sn-Mo structure is schematically presented in Scheme 1.

|
Download:
|
| Scheme 1. The synthetic strategy for the Sn-doped MoO3 metal oxide and lithium ion deintercalation mechanism. | |
{kind=link}
The prepared β-SnMoO4 has flake-like morphology that are randomly arranged about one another. The thickness of each plate was obtained approximately less than 20 nm. This layered morphology indicates lithium ions can easily transfer, therefore theoretically would deliver good dynamic performance when used as anode for LIBs.
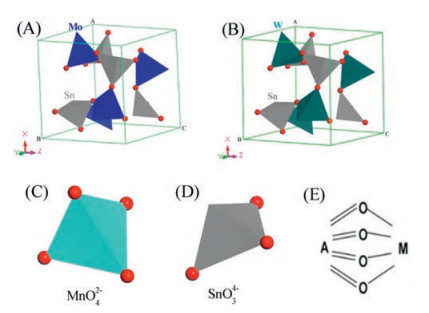
From this experiment, a temperature of 225 ℃ was chosen to obtain the nanostructured composite through observing the crystallite growth of the β-SnMoO4. DFT theory was used to simulate the structure of β-SnMoO4. The unit cells for the structure types of interest are shown in Fig. 1. According to the previous reports, β-SnMoO4 and β-SnWO4 are the similar structures, which may be expected for the combination of a cation by tetrahedral coordination and a larger cation adopting eightfold coordination [30]. Fig. 1A shows the theoretical crystal structure of β-SnMoO4 after structural optimization by Density Functional Theory. As shown in the literature [31], the calculated lattice constant of β-SnMoO4 (7.543 Å) is close to that of cubic β-SnWO4 (7.584 Å [32]) (Fig. 1B), which is consistent with results that Mo (Ⅵ) and W(Ⅵ) ions have similar ionic radium [33, 34]. Therefore, it is reasonable to conclude that β-SnMoO4 is composed of MoO42− tetrahedrons (Fig. 1C) and SnO34− trigonal pyramids (Fig. 1D) that are corner shared. As we know, Sn atoms are coordinated by three O atoms, which the Sn-O bond length is almost the same as that in β-SnWO4. The Mo atom shares the same coordination environment as the W atom which is tetrahedrally coordinated with the oxygen atoms to form corner-shared MoO4 tetrahedra [35].

|
Download:
|
| Fig. 1. Structure depiction of the (A) β-SnMoO4, (B) β-SnWO4 structure, where the transition metal ions are represented as tetrahedral shapes, the oxygen ions as red spheres. The diagrams to the below show the coordination environment of the cations, Mo6+ (C), Sn2+ (D), respectively. Both structures obey Pauling's fifth rule [34] (the rule of parsimony) and Brown's rule of maximal symmetry [35], as illustrated in the bond graph shown in Fig. 1E. The sketch is from the ref. [30]. | |
{kind=link}
The crystalline structure and quality of the as-synthesized β-SnMoO4 treated at 498 K were investigated by XRD analysis, as shown in Fig. 2A. The appearance of main distinctive diffraction peaks in the 2θ ranges at 24.50°, 27.44°, 30.14°and 46.82° was observed confirmed that the XRD peak positions are in good agreement with the crystallographic data previously reported [35], indicating the formation of a well crystalline single-phase structure. No other sharp peaks can be indexed to other impurities, which confirms that β-SnMoO4 with relatively high crystal purity have been obtained. However, there is no standard PDF card which shows the XRD profile that calculated from the optimized crystal structure for this substance by now. Therefore, the XRD profile (Fig. 2B) of β-SnMoO4 were analyzed by the Rietveld method using the XRD profile of the sample and the calculated PDF card obtained by Rietveld analysis (Fig. S1 in Supporting information). Through rigorous comparison, the XRD profile of the sample corresponds to the fact in Fig. 2B, the peaks show some crystalline nature and refined structural parameters for β-SnMoO4. The X-ray diffraction pattern (Fig. 2A) for the product shows diffraction peaks at 23.95°, 26.96°, 29.65°, 45.49°, indexed to the (200), (210), (211) and (231) planes of β-SnMoO4, respectively, which is consistent with β-SnMoO4 simulated pattern (Fig. S1).

|
Download:
|
| Fig. 2. XRD profiles and crystal structure analysis of as-synthesized β-SnMoO4: (A) XRD profile of β-SnMoO4 prepared at 498 K (red cross). (B) the calculated profiles obtained by Rietveld analysis; solid green line corresponds to the calculated intensities. Vertical ticks indicate the positions of the Bragg reflections. (C) Rietveld refinement analysis of β-SnMoO4 sample in different temperature from 573 K to 973 K. (D) XRD pattern of sample obtained at 973 K. | |
{kind=link}
Meanwhile, the XRD patterns (Fig. 2C) reveal that rutile-like SnO2/MoO2 is obtained after annealing sample in N2 from 573 K to 973 K, which matched well with reference [35]. All the peaks can be indexed to SnO2 (JCPDS card No. 41-1445) and MoO2 (JCPDS card No. 04-0673) (Fig. 2C). Obviously, the experimental spectrum of the sample heated at 673 K can be assigned entirely to Sn (Ⅳ) oxides, which is consistent with the XRD peaks in Fig. 2C that are identified as a solid solution of SnO2-MoO2 [36, 37], and finally the mixture of rutile-type SnO2 and monoclinic MoO2 were formed via the synthesis conditions in ref. [38] (i.e., 973 K for 2 h in an inert atmosphere), as shown in Fig. 2D.
To give more insights on the β-SnMoO4 sample treated at 498 K, further analyses were carried out. The topography of the sample was investigated by SEM and TEM. The result obviously shows that the prepared β-SnMoO4 has flake-like morphology with smooth surface that are randomly arranged about one another. The thickness of each plate was obtained approximately less than 20 nm (Figs. 3A–D). This layered morphology indicates lithium ions can easily transfer, therefore theoretically would deliver good dynamic performance when used as anode for LIBs. The in-depth morphological features were investigated by TEM analysis, as can be seen in Fig. 3E, indicating the layered accumulation, which may be a single layer of material at the edge. Detailed SAED (Fig. 3F) characterization on a typical nanoflake reveals that the single-crystalline material grows along the (211) direction; The atomic spacings of 3.324, 3.026 and 1.973 Å agree well with the interplanar spacings of the (210), (211) and (231) planes of the sample, respectively, which is consistent with simulated calculation. In addition, the elemental composition of as-synthesized β-SnMoO4 was identified by X-ray spectroscopy analysis (EDS, Fig. S2 in Supporting information). The EDS spectrum shows quantitative results of Sn, Mo, and O with the ratio of 1:2:8, and no any other significant impurities, this is in good accordance with the experimental protocols.

|
Download:
|
| Fig. 3. SEM images (A–D), TEM images (E) and SAED pattern (F) of the β-SnMoO4 sample nanoflake. | |
{kind=link}
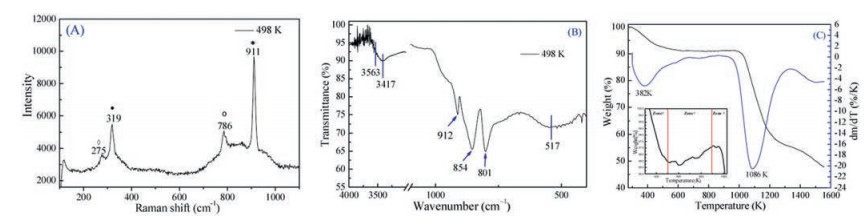
The laser Raman spectra of the β-SnMoO4 are shown in Fig. 4A, we did get several noticeable peaks observed at 911, 786, 820, 319 and 275 cm−1. As there is no report available for β-SnMoO4 nanomaterials, we compared our data with MoO3 and MoO2 nanomaterials [39, 40]. According to group theory calculations, molybdate structure exhibits (Raman and Infrared) vibrational modes (Eqs. S1 and S2 in Supporting information). A Raman spectrum with sharp peaks indicate characteristics with structural order at short-range [41]. Many factors would produce characteristic changes such as preparation methods, low temperature of synthesis, geometry or crystal size. The β-SnMoO4 crystal lattice is characterized by bonds with a covalent character between the O–Sn–O and O–Mo–O, respectively. Thus, we expect that the crystal bonds have a covalent nature in the lattice due to [SnO3] clusters [42] and [MoO4] clusters with symmetric stretching vibrations. The positions of each Raman-active mode are labeled in Table S1 (Supporting information). Slight shifts in these positions can be correlated to the degree of structural order or distortion of the lattice induced by different conditions. Also, it is possible that the degree of interaction and variations in the bond lengths induce shifts in the Raman peak positions with temperature increasing [43] (Fig. S3 in Supporting information). Increasing the temperature to 973 K results in decomposition to SnO2 and MoO2 [44, 45], resulting in sharp peaks appearing at 673, 292 cm−1, which are assigned to the bending and stretching frequencies of the nanomaterials model.

|
Download:
|
| Fig. 4. Raman spectra (A), FT-IR spectra (B) and TGA-DTA curves (C) of as-synthesized β-SnMoO4 nanomaterials. | |
{kind=link}
The FT-IR spectrum of the β-SnMoO4 is shown as Fig. 4B. It is known that the peak region 3100–3750 cm−1 is mainly due to the stretching vibration of OH groups. In the lower region, four intense peaks appear at 517, 801, 854, and 912 cm−1, which are probably due to the presence of β-SnMoO4. A single broad band centered at 517 cm−1 is due to Sn–O stretching modes, which is consistent with the literature's report [46]. The resonance absorption due to MoO stretch mode vibration is reported to lie in 905 cm−1. A strong absorption peak at 912 cm−1 in our samples confirms the presence of terminal double bonds, which is a basic characteristic of a layered structure [47]. The measured IR reflectance of materials intercalated with different temperature are shown as Fig. S4 (Supporting information), we can find that synthesized temperature obviously affects the purity and crystallinity of as synthesized samples. First, when T is increased from 498 K to 773 K, which respective locations remain unaffected (refer curves c and d). Second, when T is increased to 973 K, the three characteristic resonance absorption peaks of MoO2-SnO2 enhanced, suggesting the formation of pure MoO2-SnO2 phase with higher crystallinity at this temperature. Third, in the whole region for all T values, the disappearance of 801 and 854 cm−1 peak and the simultaneous emergence of two peaks at 623 and 642 cm−1 clearly shows a relationship between them. Therefore, it is reasonable to conclude that we exhibit (Raman and infrared) vibrational modes for the pure phase of β-SnMoO4 nanomaterials. Moreover, from the XRD analysis we have observed the same general change with an increase in the annealing temperature, which further indicates the absence of any impurities phase in our synthesized SMO nanomaterials samples.
Thermal decomposition of the formation of the oxide phases was followed by thermogravimetric analysis (TGA) and differential thermal analysis (DTA) at the temperature ranges (Fig. 4C). The β-SnMoO4 sample was taken and heated up to 1573 K in air (Vh = 10 K/min). The initial weight loss of nearly 4.13% accompanied by an endothermic event was observed in zone Ⅰ (between room temperature and 497 K), because of the loss of physically absorbed water. In zone Ⅱ (497–897 K) an increase of 1.3% of the sample was observed which is interpreted as the oxidation of SnO [48]. Ramamoorthy et al. [49] reported that the transformation of SnO into SnO2 occurs at 773–873 K during heating. Finally, in the zone Ⅲ (897–1300 K) a steep decrease in the TGA profile is observed which might be due to the decomposition of the parent material. Simultaneously, in the DTA curve two peaks at 382 and 1086 K (endothermic peaks associated with weight loss) were observed. The decrease at 382 K (in the zone Ⅲ) in the TGA profile well corresponded to the DTA profile, while an exothermic peak at 1086 K was observed, corresponds to the melting of the MoO3 phase which is accompanied by salt's decomposition [50]. Therefore, to get pure d β-SnMoO4, heat-treatment temperature at the 498 K is optimized, which can effectively prevent the formation of MoO2-SnO2 solid solution impurity phase.
X-ray photoelectron spectroscopy (XPS) was used to evaluate the information about the chemical status of the as-synthesized β-SnMoO4, as shown in Fig. 5. All binding energies were corrected with the C 1s line at 284.6 eV. The overall XPS spectrum in Fig. 5A shows the coexistence of elements Mo, C, Sn and O within the as-prepared β-SnMoO4 microparticles and no other impurities were detected, which are in good agreement with EDX report. Fig. 5B is the survey spectrum ranging from 0 to 900 eV, which consists of different characteristic peaks originating from Sn 3p at 717.5, O 1s at 530.5, Sn 3d at 487.5, Mo 3p at 417.5, C 1s at 284.6, Mo 3d at 233.5, and Sn 4d at 27.5 eV [51]. High resolution scanning XPS spectra clearly confirms the Sn 3d, Mo 3d and O 1s level by using the Gaussian fitting method, as shown in Figs. 6B–D. In the case of Mo 3d spectra (Fig. 5C), the high intense strong peaks at 232.7 and 235.8 eV can be attributed to the Mo 3d5/2 and Mo 3d3/2 of the Mo6+ state, respectively. From the Fig. 6B, the peaks at 487.2 and 495.6 eV corresponds to the Sn 3d5/2 and Sn 3d3/2 spin−orbit of Sn2+, respectively [51, 52]. The binding energies in the range of 529–532 eV were attributed to the O 1s spin−orbit level of O2−oxidation state (Fig. 6D). As is known by the literature [53], the deconvolution peak at 531.0 was due to the appearance of lattice oxygen, while 532.3 eV due to adsorbed water molecules. Hence, the obtained XPS results clearly confirmed that the valence of Mo, Sn and O are +6, +2 and −2, respectively, which is very good agreement with the phase structure of β-SnMoO4.

|
Download:
|
| Fig. 5. X-ray photoelectron spectra (XPS) for as-synthesized β-SnMoO4 sample. (A) shows the survey spectrum; (B) shows the high resolution scan for the Mo 3d peaks; (C) shows the high resolution scan for Sn3d peaks; (D) shows the high resolution scan for the O 1s peaks. | |
{kind=link}

|
Download:
|
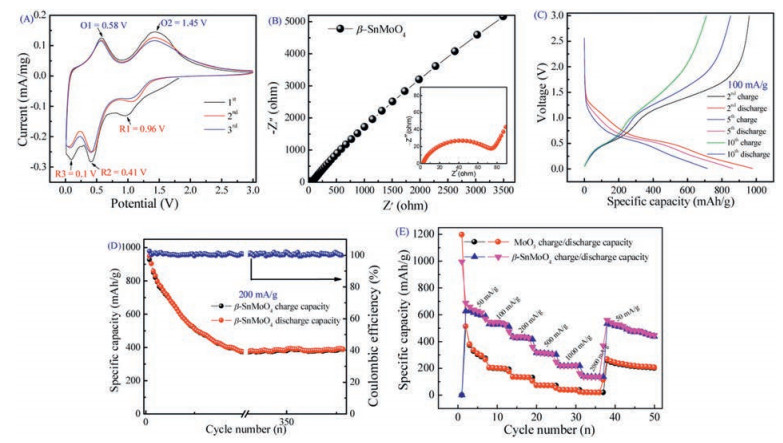
| Fig. 6. Lithium storage performances of β-SnMoO4. (A) The first three CV curves tested at a scan rate of 0.1 mV/s vs. Li/Li+. (B) Nyquist plots of the β-SnMoO4 electrode. (C, D) Cycling performance and coulombic efficiency of β-SnMoO4 tested at the current density of 100 mA/g and 200 mA/g. (E) Rate performance of β-SnMoO4 and MoO3 tested at current densities varying from 50, 100, 200, 500, 1000 and 2000, back to 50 mA/g. | |
{kind=link}
The relationship between annealing temperature and structure stability was further studied as shown in Fig. S5 (Supporting information). As the annealing temperature increasing, the weak doublet peaks at the binding energies of 231.8 and 234.9 eV can be ascribed to Mo5+ 3d5/2 and 3d3/2 [54-56]. Besides, the value of 3d doublet for Sn is consistent with those reported for SnO2 and SnO (Fig. S5b) [57, 58]. Therefore, it is reasonable to conclude that β-SnMoO4 treated at 498 K phase structure has a stable valence. Meanwhile, the valence disorders while the temperature rising, eventually leading to phase transition, which is not conducive to the formation of pure phase, may affecting its electrochemical performance.
The electrochemical performance of the β-SnMoO4 flake-like structures as the LIB anode were evaluated by CV and galvanostatic cycling. CV tests (Fig. 6) were performed in the potential window of 0.01–3 V vs. Li+/Li at a scan rate of 0.1 mV/s at room temperature.
As shown in Fig. 6a, three reduction peaks are observed in the initial cycle. The cathodic peak located at around 0.96 V (R1) is obtained from the sample during the negative scan. The reduction peak tends to Li intercalations into the electrode (Reaction 1). It can be assigned to the irreversible lithium insertion into the crystal structure (the intralayer of [MoO6]), which may cause unrecoverable structure transformation of β-SnMoO4 [59]. After that, the subsequent peaks at around 0.41 (R2) and 0.1 V (R3) might correspond to the conversion of Mo(+6) to Mo(0) and Sn(+2) to Sn(0) (Reaction 2), accompanied by the decomposition of the electrolyte and formation of SEI film [60]. During the first positive scan, the peak located at 0.58 V (O1) is indexed with the transformation of metallic Mo to MoO2 (Reaction 3); the O peak located at 1.45 V reflects the overlap of the conversions of MoO2 to MoO3 (Reaction 4) and Sn(0) to SnO2 (Reaction 5) [61].
In addition, the subsequent two CV curves almost overlap, which might show highly reversible lithium storage and repeatability during cycling [62]. Two anodic peaks are observed at 0.58 V and 1.45 V. The first peak is assigned to the oxidation of Mo to Mo4+, while the second peak is due to the oxidation of Mo4+ to Mo6+, as well as the oxidation of Sn to Sn4+ [63].

|
(1) |

|
(2) |

|
(3) |

|
(4) |

|
(5) |
Electrochemical impedance spectroscopy (EIS) is carried out to investigate the kinetic behaviors of the β-SnMoO4 electrodes. The radius of the semicircle indicates the charge transfer resistance (Rct) at the electrolyte/electrode interface [55]. Nyquist plot is constituted of a depressed semicircle in the high-frequency region and a slope line in the low-frequency region (Fig. 6B). The Rct value for β-SnMoO4 calculated to be 73 Ω, which is lower than that of pristine MoO3 (94 Ω) (Fig. S6 in Supporting information), which indicates that the prepared β-SnMoO4 could enable much easier charge transfer at the electrode interface and decrease the overall battery internal resistance. As we all know, MoO3 has poor ionic and electronic conductivity, which limits its electrochemical performance and Sn has shown a considerable volume change during the charge–discharge process that resulted in the pulverization of the anode suggesting its poor cyclic stability. The as-synthesized β-SnMoO4 has layered structure, which is able to act as a temporary support for several ions and facilitates rapid insertion and deintercalation of lithium ions and improves the stability of Sn to some extent.
In addition, the discharge/charge curves of β-SnMoO4 in the voltage range of 0.01–3 V vs. Li+/Li at a current density of 100 mA/g are displayed in Fig. 6C. The initial discharge and charge capacities are 986 and 958 mAh/g, respectively. The initial capacity loss commonly results from SEI layer formation [64]. After 20 cycles, the discharge and charge retain 515 and 510 mAh/g, respectively; even 300 cycles later, the subsequent discharge and charge curves exhibit almost no change, revealing the better cycling stability of the β-SnMoO4 sample. As the Fig. 6D shows, the initial Coulombic efficiency is nearly 100% at a current density of 200 mA/g. Furthermore, the rate capability of the electrode was tested at various current densities. The electrode can deliver highly reversible average specific capacities of 688, 162, and 560 mAh/g at the current densities of 50, 2000 and 50 mA/g, respectively Fig. 6E. Remarkably, the β-SnMoO4 delivers higher capacity and better rate performance than MoO3. The excellent electrochemical performance of β-SnMoO4 indicates that the structure can improve electrical/ionic transport in the electrode, which leads to fast electrochemical kinetics.
In summary, a novel molybdenum-based metal oxide β-SnMoO4 nanoflake was successfully fabricated by a facile one-step solid state reaction synthesis, along with post annealing. It has been demonstrated that the β-SnMoO4 be provide with typical nanoflake-like morphology with smooth surface, which is a new structure for tin molybdate that have been not reported before. The possible structure was investigated by DFT, XRD, Raman, FT-IR, SEM, TEM, SAED, and XPS analysis. For lithium storage, it for the first time delivers a discharge capacity of 395 mAh/g at the current densities of 200 mA/g, retaining 392 mAh/g after 300 cycles and high coulombic efficiency. The results indicate that the β-SnMoO4 composite is a promising anode material for LIBs and delivers higher capacity and better rate performance.
AcknowledgmentsThe authors acknowledge the support of National Key Research; Development Program of China (No. 2017YFB0102900); National Key Research and Development Plan (No. 2017YFB0102200); the Shanghai Committee of Science and Technology, China (No. 17010500500) and the Shanghai Municipal Education Commission (Peak Discipline Construction program).
| [1] |
S. Chu, A. Majumdar, Nature 488 (2012) 294. DOI:10.1038/nature11475 |
| [2] |
F. Wu, S. Zhang, B. Xi, et al., Adv. Energy Mater. 8 (2018) 1703242. |
| [3] |
H.B. Zhao, C.J. Hu, H.W. Cheng, et al., Sci. Rep. 6 (2016) 25809. DOI:10.1038/srep25809 |
| [4] |
X. Deng, Z. Wei, C. Cui, et al., J. Mater. Chem. A 6 (2018) 4013-4022. DOI:10.1039/C7TA11301C |
| [5] |
M.A. Khan, H. Zhao, W. Zou, et al., Electrochem. Energy Rev. 1 (2018) 483-530. DOI:10.1007/s41918-018-0014-z |
| [6] |
H. Zhao, L. Pan, S. Xing, et al., J. Power Sources 222 (2013) 21-31. DOI:10.1016/j.jpowsour.2012.08.036 |
| [7] |
J. Zhang, W. Lv, D. Zheng, et al., Adv. Energy Mater. 8 (2018) 1702395. |
| [8] |
J. Lu, Z. Chen, F. Pan, et al., Electrochem. Energy Rev. 1 (2018) 35-53. DOI:10.1007/s41918-018-0001-4 |
| [9] |
Y. Xie, L. Fang, H. Cheng, et al., J. Mater. Chem. A 4 (2016) 15612-15620. DOI:10.1039/C6TA06164H |
| [10] |
H. Shi, L. Wei, Z. Chen, et al., Adv. Funct. Mater. 28 (2018) 1800508. DOI:10.1002/adfm.201800508 |
| [11] |
J. Ma, F. Li, Z. Wei, et al., J. Mater. Chem. A 7 (2019) 9406-9431. DOI:10.1039/C8TA11999F |
| [12] |
Y. Fang, R. Hu, B. Liu, et al., J. Mater. Chem. A 7 (2019) 5363-5372. DOI:10.1039/C8TA12069B |
| [13] |
L. Mai, L. Xu, C. Han, et al., Nano Lett. 10 (2010) 4750. DOI:10.1021/nl103343w |
| [14] |
V. Etacheri, R. Marom, E. Ran, et al., Energy Environ. Sci. 4 (2011) 3243-3262. DOI:10.1039/c1ee01598b |
| [15] |
S. Qi, D. Wu, Y. Dong, et al., Chem. Eng. J. 370 (2019) 185-207. DOI:10.1016/j.cej.2019.03.166 |
| [16] |
J.C. Chou, Y.L. Chen, M.H. Yang, et al., J. Mater. Chem. A 1 (2013) 8753-8758. DOI:10.1039/c3ta11027c |
| [17] |
J. Hong, Y.W. Lee, B. Hou, et al., ACS Appl. Mater. Interfaces 8 (2016) 35227-35234. DOI:10.1021/acsami.6b11584 |
| [18] |
J. Huang, Z. Wei, J. Liao, et al., J. Energy Chem. 33 (2019) 100-124. DOI:10.1016/j.jechem.2018.09.001 |
| [19] |
B. Das, M.V. Reddy, C. Krishnamoorthi, et al., Electrochim. Acta 54 (2009) 3360-3373. DOI:10.1016/j.electacta.2008.12.049 |
| [20] |
Y. Liang, Q. Liu, A.M. Asiri, X. Sun, Electrochim. Acta 153 (2015) 456-460. DOI:10.1016/j.electacta.2014.11.193 |
| [21] |
L. Wan, J. Shen, Y. Zhang, X. Li, J. Alloys. Compd. 708 (2017) 713-721. DOI:10.1016/j.jallcom.2017.03.078 |
| [22] |
X. Mu, Y. Zhang, H. Wang, et al., Electrochim. Acta 211 (2016) 217-224. DOI:10.1016/j.electacta.2016.06.072 |
| [23] |
J. Liu, J. Liang, C. Wang, J. Ma, J. Energy Chem. 33 (2019) 160-166. DOI:10.1016/j.jechem.2018.09.006 |
| [24] |
A. Belsky, M. Hellenbrandt, V.L. Karen, P. Luksch, Acta Crystallogr. 58 (2002) 364-369. DOI:10.1107/S0108768102006948 |
| [25] |
I. Shakir, M. Shahid, M. Nadeem, D.J. Kang, Electrochim. Acta 72 (2012) 134-137. DOI:10.1016/j.electacta.2012.04.002 |
| [26] |
W. Geng, X. Zhao, H. Liu, X. Yao, J. Phys. Chem. C 117 (2013) 10536-10544. DOI:10.1021/jp401733h |
| [27] |
J. Xu, Z. Xue, N. Qin, et al., Sens. Actuators B-Chem. 242 (2017) 148-157. DOI:10.1016/j.snb.2016.09.193 |
| [28] |
J.P. Perdew, K. Burke, M. Ernzerhof, Phys. Rev. Lett. 77 (1996) 3865-3868. DOI:10.1103/PhysRevLett.77.3865 |
| [29] |
O.I. Lebedev, F. Millange, C. Serre, et al., Chem. Mater. 17 (2005) 6525-6527. DOI:10.1021/cm051870o |
| [30] |
M.W. Stoltzfus, P.M. Woodward, R. Seshadri, et al., Inorg. Chem. 46 (2007) 3839-3850. DOI:10.1021/ic061157g |
| [31] |
W. Jeitschko, A.W. Sleight, Acta Crystallogr. 28 (2010) 3174-3178. |
| [32] |
I.S. Cho, C.H. Kwak, W.K. Dong, et al., J. Phys. Chem. C 113 (2009) 10647-10653. DOI:10.1021/jp901557z |
| [33] |
R.D. Shannon, Acta Crystallogr. 32 (1976) 751-767. DOI:10.1107/S0567739476001551 |
| [34] |
N.C. Jayadevan, K.D.S. Mudher, D.M. Chackraburtty, Acta Crystallogr. 31 (1975) 2277-2280. DOI:10.1107/S056774087500739X |
| [35] |
H. Hayashi, S. Katayama, T. Komura, et al., Adv. Sci. 4 (2017) 1600246. DOI:10.1002/advs.201600246 |
| [36] |
J. Pannetier, J. Bassasalsina, J. Rodriguezcarvajal, V. Caignaert, Nature 346 (1990) 343-345. DOI:10.1038/346343a0 |
| [37] |
G.H. Rao, I.D. Brown, Acta Crystallogr. 54 (1998) 221-230. DOI:10.1107/S0108768197013426 |
| [38] |
A.M.E.S. Raj, C. Mallika, O.M. Sreedharan, K.S. Nagaraja, Mater. Res. Bull. 36 (2001) 837-845. DOI:10.1016/S0025-5408(01)00570-0 |
| [39] |
S.H. Lee, Y.H. Kim, R. Deshpande, et al., Adv. Mater. 20 (2008) 3627-3632. DOI:10.1002/adma.200800999 |
| [40] |
F. Rullens, N. Deligne, A. Laschewsky, M. Devillers, J. Mater. Chem. 15 (2005) 1668-1676. DOI:10.1039/b418233b |
| [41] |
Z.C. Ling, H.R. Xia, D.G. Ran, et al., Chem. Phys. Lett. 426 (2006) 85-90. DOI:10.1016/j.cplett.2006.05.093 |
| [42] |
R.D.M. Bomio, Batista, et al., Polyhedron 50 (2013) 532-545. DOI:10.1016/j.poly.2012.12.001 |
| [43] |
J.X. Zhou, M.S. Zhang, J.M. Hong, Z. Yin, Solid State Commun. 138 (2006) 242-246. DOI:10.1016/j.ssc.2006.03.007 |
| [44] |
J. Ni, Y. Zhao, L. Li, L. Mai, Nano Energy 11 (2015) 129-135. DOI:10.1016/j.nanoen.2014.10.027 |
| [45] |
S.R. Ede, S. Kundu, ACS Sustain. Chem. Eng. 3 (2015) 2321-2336. DOI:10.1021/acssuschemeng.5b00627 |
| [46] |
J. Li, H. Li, Z. Wang, et al., J. Power Sources 107 (2002) 1-4. DOI:10.1016/S0378-7753(01)00964-8 |
| [47] |
T.S. Sian, G.B. Reddy, Appl. Surf. Sci. 236 (2004) 1-5. DOI:10.1016/j.apsusc.2004.03.259 |
| [48] |
H. Yang, Y. Hu, A. Tang, et al., J. Alloys. Compd. 363 (2004) 276-279. DOI:10.1016/S0925-8388(03)00473-0 |
| [49] |
R. Ramamoorthy, M.K. Kennedy, H. Nienhaus, et al., Sens. Actuators B-Chem. 88 (2003) 281-285. DOI:10.1016/S0925-4005(02)00370-2 |
| [50] |
J. Lalande, R. Ollitrault-Fichet, P. Boch, J. Eur. Ceram. Soc. 20 (2000) 2415-2420. DOI:10.1016/S0955-2219(00)00153-9 |
| [51] |
S.A. Suthanthiraraj, Y.D. Premchand, Ionics 10 (2004) 254-257. DOI:10.1007/BF02382825 |
| [52] |
M. Martos, J. Morales, L. Sánchez, J. Mater. Chem. 12 (2002) 2979-2984. DOI:10.1039/B203993A |
| [53] |
R. Karthik, K.J. Vinoth, S.M. Chen, et al., ACS Appl. Mater. Interfaces 9 (2017) 26582-26592. DOI:10.1021/acsami.7b06851 |
| [54] |
J.F. Moulder, W.F. Stickle, P.E. Sobol, et al., Chem. Phys. Lett. 220 (1995) 7-10. |
| [55] |
Y. Yao, N. Xu, D. Guan, et al., ACS Appl. Mater. Interfaces 9 (2017) 39425-39431. DOI:10.1021/acsami.7b13529 |
| [56] |
Y. Hosogi, Y. Shimodaira, H. Kato, et al., Chem. Mater. 20 (2008) 1299-1307. DOI:10.1021/cm071588c |
| [57] |
B. Mendoza-Sánchez, T. Brousse, C. Ramirez-Castro, et al., Electrochim. Acta 91 (2013) 253-260. DOI:10.1016/j.electacta.2012.11.127 |
| [58] |
J. Światowskamrowiecka, S.D. Diesbach, V. Maurice, et al., J. Phys. Chem. C 112 (2016) 11050-11058. |
| [59] |
Y. Dong, S. Li, H. Xu, et al., Phys. Chem. Chem. Phys. 15 (2013) 17165-17170. DOI:10.1039/c3cp53267d |
| [60] |
Z. Yin, Y. Xiao, X. Wang, et al., Nanoscale 8 (2015) 508-516. |
| [61] |
Z. Ju, E. Zhang, Y. Zhao, et al., Small 11 (2015) 4753. DOI:10.1002/smll.201501294 |
| [62] |
J. Meng, Z. Liu, C. Niu, et al., Mater. Horiz. 5 (2017) 78-85. |
| [63] |
C. Han, X. Ren, Q. Li, et al., Nano Res. 11 (2018) 1-9. DOI:10.1007/s12274-017-1616-1 |
| [64] |
J. Yao, Y. Gong, S. Yang, et al., ACSAppl. Mater. Interfaces 6 (2014) 20414-20422. DOI:10.1021/am505983m |