 2020, Vol. 31
2020, Vol. 31 

Marine bioresources, especially marine microorganisms, are an irreplaceable and attractive research area in drug discovery, which has successfully caught considerable attention from natural product chemists and pharmacologists. As time goes by, a large number of marine microbial secondary metabolites with unique chemical backbones and remarkable bioactivities are being reported, including terpenes [1], alkaloids [2], polyketides [3], etc., some of which show antibacterial, antiviral, antioxidant, antiallergic activities and so on. The discovery of these compounds provides outpouring lead molecules for innovative drug research.
Cyclopianes represent a rare class of tetracyclic diterpenes defined by a highly fused and rigid 6/5/5/5 ring system, whose absolute structures have never been reported by X-ray crystallography experiment until 2018 when Abe and co-workers first confirmed this skeleton with the aid of the crystal sponge method [4]. Interestingly, cyclopiane diterpenes exhibited excellent bioactivities, such as potent conidia inducing activity [5], selective cytotoxicity against human leukemia (HL-60) cell, and antibacterial activity against methicillin-resistant Staphylococcus aureus (MRSA) [6]. Inspired by their complex chemical architectures and promising bioactivities, we performed a systematic investigation on the secondary metabolites of a sea sediment-derived fungus Penicillium sp. TJ403-2. As a result, three new rare diterpenes (1–3) and thirteen known compounds (4–16) (Fig. 1) were characterized. The isolation, structure elucidation, and antiinflammatory activity of these new compounds are reported in this paper.

|
Download:
|
| Fig. 1. Chemical structures of compounds 1–16. | |
The fungal strain Penicillium sp. TJ403-2 was cultured on PDA plates at 28℃ for 7 days, and then inoculated into 100×1000 mL sterile Erle nmeyer flasks. Each flask contained 250g rice and 250 mL 3% sea salt distilled water. After 25 days of culture at room temperature, the fermented materials of Penicillium sp. TJ403-2 were extracted with EtOAc for six times to obtain a crude extract (166g).
The EtOAc extract (166g) was chromatographed on silica gel CC using an increasing gradient of petroleum ether (PE)-ethyl acetate (EtOAc) (30:1, 20:1, 10:1, 5:1, 2:1, 1:1, 0:1) to obtain seven main fractions (A–G).
Fraction B (14g) was loaded on an RP-C18 column eluted with MeOH–H2O (20%–100%) to get five fractions (B1–B5). Fraction B2 (1.48g) was subjected to silica gel CC (PE–EtOAc, 4:1–1:1) to give three main subfractions (B2.1–B2.3). Fraction B2.1 was applied to Sephadex LH-20, eluted with CH2Cl2–MeOH (1:1, v/v), and further purified by RPHPLC [CH3CN–H2O, 54:46, 2.0 mL/min, 210 nm, tR=15 min for 3 (3.8 mg), tR=21 min for 4 (33.7 mg); MeOH–H2O, 64:36, 2.0 mL/min, 210 nm, tR=25 min for 12 (6.1 mg), tR=29 min for 13 (6.3 mg)]. Fraction B2.2 was fractionated on Sephadex LH-20 eluted with CH2Cl2–MeOH (1:1, v/v), and further purified by RPHPLC [CH3CN–H2O, 52:48, 2.0 mL/min, 210 nm, tR=24 min, for 1 (5.7 mg); MeOH–H2O, 65:35, 2.0 mL/min, 210 nm, tR=32 min for 16 (4.1 mg); CH3CN–H2O, 56:44, 2.0 mL/min, 210 nm, tR=19 min for 10 (8.7 mg)]. Fraction B3 was loaded on silica gel CC (PE–EtOAc, 5:1–1:1), followed by Sephadex LH-20 (CH2Cl2–MeOH, 1:1, v/v), and further purified by RPHPLC [MeOH–H2O, 70:30, 2.0 mL/min, 210 nm, tR=51 min for 5 (4.2 mg), tR=53 min for 6 (5.2 mg), tR=55 min for 7 (8.5 mg)]. Fraction B4 was chromatographed on silica gel CC and then purified by RPHPLC [CH3CN–H2O, 75:25, 2.0 mL/min, 210 nm, tR=28 min for 8 (18.7 mg), tR=32 min for 9 (8.9 mg)].
Fraction C (10g) was subjected to an RP-C18 column eluted with MeOH–H2O (20:80–100:0) to obtain four fractions (C1–C4). Fraction C2 (0.98g) was chromatographed on Sephadex LH-20 (CH2Cl2–MeOH, 1:1, v/v), and further purified by RPHPLC [CH3CN–H2O, 45:55, 2.0 mL/min, 210 nm, tR=19 min for 2 (5.5 mg); MeOH–H2O, 56:44, 2.0 mL/min, 210 nm, tR=26 min for 14 (14.1 mg), tR=35 min for 15 (17.5 mg)].
Fraction D (5g) was separated by Sephadex LH-20 (MeOH), and further purified by semi-preparative RPHPLC to yield 11 (MeOH–H2O, 56:44, 2.0 mL/min, 210 nm, tR=26 min, 14.2 mg).
13β-Hydroxy conidiogenone C (1): Colorless block crystals; [α]D25: +3.46 (c 1, MeOH); UV (MeOH) λmax (logε)=231 (3.96) nm; ECD (MeOH) λmax (Δε)=234 (+2.77) nm; IR (KBr): νmax 3431, 2950, 2873, 1657, 1456, 1383, 1266, 1069, 1027 cm–1; 1H and 13C NMR data are shown in Table 1; HRESIMS: m/z 341.2127 ([M+Na]+), calcd. for C20H30O3Na+: 341.2087.
|
|
Table 1 1H NMR (400MHz) and 13C NMR (100MHz) data for compounds 1–3 in methanol-d4 (δ in ppm, J in Hz). |

12β-Hydroxy conidiogenone C (2): Colorless block crystals; [α]D25: –0.22 (c l, MeOH); UV (MeOH) λmax (logε)=230 (4.05) nm; ECD (MeOH) λmax (Δε)=237 (+3.46) nm; IR (KBr): νmax 3413, 2956, 1645, 1465, 1378, 1087, 1025, 831 cm–1; 1H and 13C NMR data are shown in Table 1; HRESIMS: m/z 341.2093 ([M+Na]+), calcd. for C20H30O3Na+: 341.2087.
12β-Hydroxy conidiogenone D (3): Colorless block crystals; [α]D25: +3.22 (c l, MeOH); UV (MeOH) λmax (logε)=231 (3.94) nm; ECD (MeOH) λmax (Δε)=243 (+1.15) nm; IR (KBr): νmax 3432, 2956, 2871, 1649, 1461, 1379, 1055 cm–1; 1H and 13C NMR data are shown in Table 1; HRESIMS: m/z 341.2084 ([M+Na]+), calcd. for C20H30O3Na+: 341.2087.
Compound 1 was obtained as colorless block crystals, and its molecular formula was deter mined to be C20H30O3, by the analysis of HRESIMS data at m/z 341.2127 [M+Na]+ (calcd. for C20H30O3Na+: 341.2087), corresponding to six degrees of unsaturation. The IR spectrum shows an absorption band for the OH group (3413 cm–1). By analysis of its 1H, 13C and DEPT NMR data (Table 1) and in association with the HSQC spectrum, 20 carbons resonances were observed, including four methyls (δH 1.25/δC 19.1, δH 1.07/δC 20.5, δH 1.17/δC 21.6, δH 1.35/δC 33.5), five sp3 methylenes (δC 35.2, 39.9, 48.5, 49.9, 67.8), six methines (δH 2.73/δC 39.0, δH 2.30/δC 56.8, δH 1.81/δC 67.4, δH 4.05/δC 81.8, δH 5.96/δC 127.9, δH 7.13/δC 157.2), and five quaternary carbons (δC 50.6, 50.6, 58.8, 60.4, 208.2). Apart from two degrees of unsaturation occupied by two olefinic carbons (δC 127.9 and 157.2) and a carbonyl carbon (δC 208.2), a tetracyclic ring system should exist in compound 1.
Detailed comparison of the 1D NMR data of 1 with those of 7 (conidigenone C) [2] suggested that both compounds were structural analogues featuring a cyclopiane diterpene skeleton. The only difference was that the C-13 was hydroxylated in 1, as supported by the 1H–1H COSY correlation between H2-12 (δH 2.08, 1.69) and H-13 (δH 4.05) and HMBC correlations (Fig. S1 in Supporting information) of H3-19 with C-13 and of H-13 with C-20. In the NOESY spectrum, the NOE correlations (Fig. S1) of H-13 (δH 4.05)/H-4α (δH 2.73), H-13 (δH 4.05)/H3-19α (δH 1.07) suggested that OH-13 was β-oriented. After repeated recrystallization with two-phase solvents, a suitable crystal of 1 was obtained and then subjected to single-crystal X-ray diffraction analysis with Cu Kα (Fig. 2) [Flack parameter=0.03 (3)], which not only confirmed its planar structure but also suggested the absolute stereochemistry of 1 to be 4S, 5S, 6S, 9R, 11S, 13S, 14S, 15S, and it was named 13β-hydroxy conidiogenone C.

|
Download:
|
| Fig. 2. ORTEP drawings of compounds 1–3. | |
Compound 2 was also obtained as colorless block crystals. According to the HRESIMS data at m/z 341.2093 [M+Na]+ (calcd. for C20H30O3Na+: 341.2087), its molecular formula was confirmed to be C20H30O3. By comparing its 1H, 13C and DEPT NMR data (Table 1), it can be found that Compound 2 is an analogue of known Compound 7 (conidigenone C) [2]. The HMBC correlations (Fig. S1) of H-12 with C-10 and of H3-18 with C-12 indicated the only difference between 2 and 7 was that C-12 was hydroxylated in 2.
The relative configuration of Compound 2 was deter mined by the NOESY spectrum. Key NOE correlations (Fig. S1) of H-12 (δH 4.14)/H-6α (δH 2.43), H-12 (δH 4.14)/H3-19α (δH 1.01) suggested that OH-12 was β-oriented. The absolute configuration of Compound 2 was deter mined to be 4S, 5S, 6S, 9R, 11R, 12R, 14S, 15R by comparing its ECD spectrum (Fig. S2 in Supporting information) with that of Compound 1 to show the identical Cotton effects. In addition, the planar structure and absolute configuration of 2 were further confirmed by single-crystal X-ray crystallography experiment (Fig. 2) [Flack parameter=0.046(19)], and it was named 12β-hydroxy conidiogenone C.
Compound 3 possessed a molecular formula of C20H30O3, as established by its HRESIMS data at m/z 341.2084 [M+Na]+ (calcd. for C20H30O3Na+: 341.2087). The 1D NMR data (Table 1) of 3 were similar to the known Compound 8 (conidiogenone D) [2], differing in that Compound 3 had one more hydroxyl group than 8. Comparison of their 2D NMR data suggested that the CH2-12 in 8 was hydroxylated in 3. In the NOESY spectrum (Fig. S1), the correlations of H-12 (δH 4.11)/H-4α (δH 2.87) and H-12 (δH 4.11)/H-19α (δH 3.37) suggested that OH-12 was β-oriented in 3. The similar ECD spectra (Fig. S2) of 2 and 3 indicated that the absolute configuration of 3 should be 4S, 5S, 6S, 9R, 11R, 12R, 14R, 15R, which was further confirmed by the single-crystal X-ray crystallographic analysis (Fig. 2) [Flack parameter = –0.08 (10)], and this compound was named 12β-hydroxy conidiogenone D.
Thirteen already known compounds were identified as conidiogenone I (4) [6], conidiogenone E (5) [2], conidiogenone F (6) [2], conidiogenone C (7) [2], conidiogenone D (8) [2], (3S, 7R)-7-hydroxyresorcylide (9) [7], (3S, 7S)-7-hydroxyresorcylide (10) [7], (3R, 7R)-3, 4, 5, 6, 7, 8-hexahydro-7, 12, 14-trihydroxy-3-methyl-1H-2-benzoxacyclodode (11) [8], (3R, 7S)-3, 4, 5, 6, 7, 8-hexahydro-7, 12, 14-trihydroxy-3-methyl-1H-2-benzoxacyclododecin-1, 9(10H)-dione (12) [8], meleagrin (13) [9], LL-S490β (14) [10], methyl N-(phenylacetyl)-l-tryptophanate (15) [11], and isocitreohybridone B (16) [12] by comparison of their NMR and HRESIMS data with the literature.
In our continuing endeavors to discover antiinflammatory agents from natural products [13-18], all of the new isolates were assessed for their inhibitory effects on LPS-induced NO production in RAW 264.7 cells at nontoxic concentrations. Among these tested compounds, Compound 1 markedly decreased LPS-induced NO production with an IC50 value of 2.19±0.25 μmol/L, which was even three fold lower than that of indometacin (Table 2).
|
|
Table 2 Inhibitory activities of tested compounds on NO production. |

{kind=link}
{kind=link}
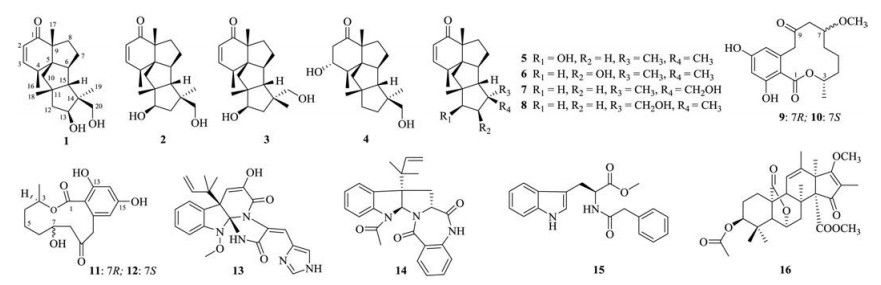
Many inflammatory factors such as interleukins, chemokines, and cytokines are recently demonstrated to be initiators of the inflammatory response and mediators of the development of chronic inflammatory diseases. To fully understand the antiinflammatory effects, we exa mined the influence of Compound 1 on the production of 23 kinds of cytokines induced by LPS using suspension array technology. Compound 1 could significantly inhibit the production of IL-1β, IL-13, TNF-α, GM-CSF, MIP-1β and MCP-1 of them (Fig. 3).

|
Download:
|
| Fig. 3. Compound 1 inhibits the production of IL-1β, IL-13, TNF-α, GM-CSF, MIP-1β, and MCP-1 in LPS-activated RAW264.7 cells. The cells were pretreated for 2h with the indicated concentrations of 1 and stimulated for 12h with LPS (1μg/ mL). Each value represents the mean±SD of triplicate cultures. ##P < 0.01, ###P < 0.001, compared with the normal group; *P < 0.05, **P < 0.01 and ***P < 0.001 compared with the group treated with LPS. | |
{kind=link}
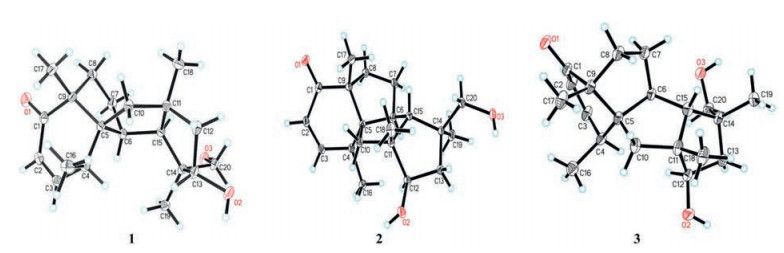
Protein expression levels of inducible nitric oxide synthesis (iNOS) and cyclooxygenase-2 (COX-2) in the RAW264.7 cells were significantly up-regulated in response to LPS, while Compound 1 suppressed the iNOS and COX-2 protein expression in a dose-dependent manner (Fig. 4A). Substantial evidence indicates that NF-κB, a well-known nuclear transcription factor, can induce transcription of several inflammatory genes such as inducible NO synthase and the pro-inflammatory cytokines. The inflammatory stimuli trigger the translocation of NF-κB to nucleus, which activates the target genes of pro-inflammatory cytokines.

|
Download:
|
| Fig. 4. (A) Effects of Compound 1 on the protein expression levels of iNOS and COX-2 in RAW264.7 cells stimulated with LPS. (B) NF-κB p65 activation level in RAW264.7 cells stimulated with LPS. The cells were pretreated for 2h with the indicated concentrations of 1 and stimulated for 12h with LPS (1μg/ mL). | |
{kind=link}
Treatment of RAW264.7 cells with LPS caused a significant increase in the nuclear translocation of p65 (RelA) subunit of NF-κB (Fig. 4B). As shown in Fig. 4, Compound 1 abolished LPS-induced nuclear translocation of NF-κB p65. In summary, we demonstrated that Compound 1 acted as a potential antiinflammatory agent to inhibit inflammatory mediators and cytokines in vitro. The downregulation of the NF-κB pathway was demonstrated to be involved in the inhibition of inflammatory mediators by Compound 1. These results provided comprehensive data for understanding the antiinflammatory effects and potential mechanisms of Compound 1.
In conclusion, three new rare diterpenes (1–3), together with thirteen known compounds (4–16), were isolated and identified from a sea sediment-derived fungus Penicillium sp. TJ403-2. Interestingly, the highly fused and strained 6/5/5/5 ring skeleton of cyclopiane diterpenes have never been reported by X-ray crystallography experiment until 2018 when Abe and co-workers first confirmed this skeleton by the crystal sponge method, and in this study we further confirmed this conclusion by single-crystal X-ray diffraction experiment. In addition, the antiinflammatory activity of cyclopiane diterpenes was first reported in this study and Compound 1 was found to show notable inhibitory potency with an IC50 value of 2.19±0.25 μmol/L, which was three fold lower than the positive control indomethacin (IC50=8.76±0.92 μmol/L). Further Western blot and immunofluorescence experiments indicated that Compound 1 could inhibit the NF-κB-activated pathway and thus provided remarkable antiinflammatory action. Our findings suggest that cyclopiane diterpenes are promising to become new antiinflammatory agents and penicillia-associated fungi have great potentials in the discovery of natural products with novel structures and pharmacological activities.
AcknowledgmentsWe thank the Analytical and Testing Center at Huazhong University of Science and Technology for measuring ECD and IR spectra. This work was financially supported by the Program for Changjiang Scholars of Ministry of Education of the People's Republic of China (No. T2016088), the National Natural Science Foundation for Distinguished Young Scholars (No. 81725021), the Innovative Research Groups of the National Natural Science Foundation of China (No. 81721005), and the National Natural Science Foundation of China (Nos. 81573316 and 21702067).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.04.036.
| [1] |
(a) B. Yang, W. Sun, J. Wang, et al., Mar. Drugs 16 (2018) 110-118; (b) L.L. Chen, F.D. Kong, P. Wang, et al., Chin. Chem. Lett. 28 (2017) 222-225. |
| [2] |
L. Du, D. Li, T. Zhu, et al., Tetrahedron 65 (2009) 1033-1039. DOI:10.1016/j.tet.2008.11.078 |
| [3] |
(a) A. Carroll, B. Copp, R. Davis, et al., Nat. Prod. Rep. 36 (2019) 122-173; (b) J. Xu, H. Tan, Y. Chen, et al., Chin. Chem. Lett. 30 (2019) 439-442. |
| [4] |
T. Mitsuhashi, T. Kikuchi, S. Hoshino, et al., Org. Lett. 20 (2018) 5606-5609. DOI:10.1021/acs.orglett.8b02284 |
| [5] |
T. Roncal, S. Cordobés, U. Ugalde, et al., Tetrahedron Lett. 43 (2002) 6799-6802. DOI:10.1016/S0040-4039(02)01493-4 |
| [6] |
S.S. Gao, X.M. Li, Y. Zhang, et al., Chem. Biodivers. 8 (2011) 1748-1753. DOI:10.1002/cbdv.201000378 |
| [7] |
L. Kuppers, W. Ebrahim, M. El-Neketi, et al., Mar. Drugs 15 (2017) 359-374. DOI:10.3390/md15110359 |
| [8] |
C.J. Barrow, J. Nat. Prod. 60 (1997) 1023-1025. DOI:10.1021/np970200x |
| [9] |
M.S. Mady, M.M. Mohyeldin, H.Y. Ebrahim, et al., Bioorg. Med. Chem. 24 (2016) 113-122. DOI:10.1016/j.bmc.2015.11.038 |
| [10] |
Y. Kimura, T. Hamasaki, H. Nakajima, et al., Tetrahedron Lett. 23 (1982) 225-228. DOI:10.1016/S0040-4039(00)86791-X |
| [11] |
X.L. Huang, Y. Gao, D.Q. Xue, et al., Helv. Chim. Acta 94 (2011) 1838-1842. DOI:10.1002/hlca.201100104 |
| [12] |
S. Kosemura, Tetrahedron 59 (2003) 5055-5072. DOI:10.1016/S0040-4020(03)00739-7 |
| [13] |
F. Li, W. Sun, J. Guan, et al., Org. Biomol. Chem. 16 (2018) 8751-8760. DOI:10.1039/C8OB02353K |
| [14] |
M. Liu, W. Sun, J. Wang, et al., Bioorg. Chem. 80 (2018) 525-530. DOI:10.1016/j.bioorg.2018.06.029 |
| [15] |
M. Liu, Q. Zhou, J. Wang, et al., RSC Adv. 8 (2018) 13040-13047. DOI:10.1039/C8RA01840E |
| [16] |
F. Li, W. Sun, J. Guan, et al., Org. Lett. 20 (2018) 7982-7986. DOI:10.1021/acs.orglett.8b03553 |
| [17] |
Z. Hu, Y. Wu, S. Xie, et al., Org. Lett. 19 (2017) 258-261. DOI:10.1021/acs.orglett.6b03557 |
| [18] |
Z. Hu, W. Sun, F. Li, et al., Org. Lett. 20 (2018) 5198-5202. DOI:10.1021/acs.orglett.8b02137 |