 2020, Vol. 31
2020, Vol. 31 


b Department of Oral and Cranio-Maxillofacial, Ninth People's Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai 200240, China;
c Department of Obstetrics, Shanghai First Maternity and Infant Hospital, Tongji University School of Medicine, Shanghai 201204, China
Resulting from the rapid emission of greenhouse gases, the natural greenhouse effect has caused a severe impact on global warming. Extensive efforts have been dedicated to the development of cost-effective technology for capturing carbon dioxide (CO2), the major greenhouse gas [1]. Adsorption of CO2 by using porous materials was considered as the most promising method for CO2 recovery. An ideal candidate material for CO2 adsorption should possess characteristics of large surface area and excellent physicochemical stability. A large amount of porous materials has been developed for CO2 adsorption [2], such as zeolites [3], metal organic frameworks (MOFs) [4, 5], and covalent organic frameworks (COFs) [6-8]. COFs which are constituted of light-weight elements bonded by covalent bonds [9, 10], are a novel class of highly designable crystalline porous organic polymers with permanent pores and periodic structures. Specifically in two-dimensional (2D) COFs, the organic building blocks are linked by covalent bonds to form extended 2D graphene-like organic sheets with polygon topology. The 2D sheets stack to constitute layered frameworks with ordered one-dimensional (1D) open channels. The shape and size of the channels are highly designable. Owing to the low skeleton density, high porosity, and high stability, COFs have exhibited potential applications in heterogeneous catalysis [11-14], gas storage and separation [6-8], energy storage [15-24], ion conduction [25].
Experimental studies have shown that COFs are promising materials for CO2 storage [6-8, 36-31]. Jiang et al. have reported their channel-wall engineering strategies for the functionalization of the channel walls with different groups to convert a conventional COF into an outstanding platform for CO2 capture via ring opening reaction and click reaction [7].
Herein, we report our novel channel-wall engineering strategy by rapid microwave-assisted anion exchange reaction based on SJTU-COF-Br, which consists of cationic framework and bromide counter anion. By using this novel method three new COFs (SJTU-COF-Cl, SJTU-COF-AcO and SJTU-COF-CF3SO3) with the same cationic framework but different counter anions (chloride, acetate or trifluomethanesulfonate) were rapidly obtained. Among them, SJTU-COF-AcO showed significantly enhanced CO2 uptake capacity, which was increased to 1.7 times as compared with the pristine SJTU-COF-Br, benefitted from the interaction between CO2 and acetate anion. For the first time, the effect of the counter anions to the CO2 capacity in the cationic COFs was investigated. The results suggest that anion exchange is a promising method to tailor the pores of COFs for high-performance CO2 capture [32, 33] (Scheme 1).

|
Download:
|
| Scheme 1. Schematic representation of the synthesis of SJTU-COFs. | |
{kind=link}
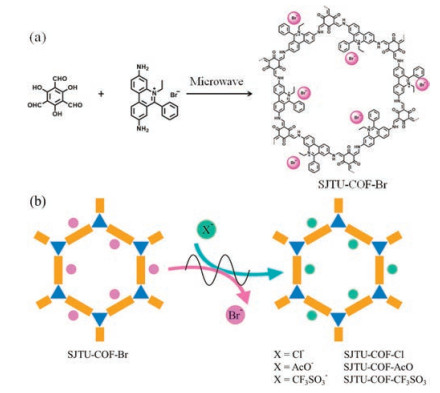
As for the synthesis of products, SJTU-COF-Br was synthesized under microwave heating conditions by preparing a 3:2 molar ratio solution of ethidium bromide (EB, 0.6 mmol) and 1, 3, 5-triformylphloroglucinol (Tp, 0.4 mmol) in a mixture of 1, 4-dioxane/mesitylene/3 mol/L acetic acid (3 mL:3 mL:1 mL). The mixture was degassed by three freeze-pump-thaw cycles and sealed under nitrogen in a 20-mL glass microwave tube. The tube was heated by microwave irradiation with a CEM Explorer microwave synthesizer at 100 ℃ for 3 h. The precipitate was collected by filtration and washed with DMSO, acetone, saturated solution of potassium bromide and deionized water. Then the powder was washed by using standard Soxhlet method with THF as solvent to remove any impurities adsorbed in the porous. The red powder was dried at 100 ℃ under vacuum overnight to yield the SJTU-COF-Br (255 mg), and the proportion of the isolated yield was 85%.
Then we obtained the SJTU-COF-Cl, SJTU-COF-AcO and SJTU-COF-CF3SO3. 500 mg of SJTU-COF-Br was dispersed in 10 mL of saturated solution of corresponding salts (sodium chloride, sodium acetate and sodium trifluomethanesulfonate). The mixture was heated by microwave irradiation at 80 ℃ with stirring for 30 min. The obtained precipitate was filtered and washed with deionized water. This anion exchange process was repeated twice. Then, the precipitate was filtered and washed with deionized water and acetone, and dried at 100 ℃ under vacuum overnight and named as SJTU-COF-Cl, SJTU-COF-AcO and SJTU-COF-CF3SO3, respectively [34, 35].
SJTU-COF-Br was synthesized by using a microwave-assisted solvothermal method (Fig. 1a), which took significantly less time than that under conventional heating conditions (3 days) [25]. SJTU-COF-Cl, SJTU-COF-AcO and SJTU-COF-CF3SO3 were prepared by rapid microwave-assisted anion exchange of SJTU-COF-Br with sodium chloride, sodium acetate or sodium trifluomethanesulfonate, respectively (Fig. 1b).

|
Download:
|
| Fig. 1. Schematic representation of (a) the microwave-assisted synthesis of SJTU-COF-Br and (b) the microwave-assisted anion exchange in SJTU-COFs. | |
{kind=link}
The bonds' formation in SJTU-COFs was investigated by FT-IR spectra (Fig. 2a). In the spectrum of SJTU-COF-Br, the disappearance of characteristic N—H stretching band of EB (3200-3400 cm-1) and C=O stretching band of Tp (1635 cm-1) indicated the completion of microwave-assisted polymerization. The keto form of SJTU-COF-Br can be indicated by the appearance of C–N and C=C stretching bands around 1252 and 1590 cm-1, respectively. Compared with the spectrum of SJTU-COF-Br, the spectra of SJTU-COF-Cl, SJTU-COF-AcO, and SJTU-COF-CF3SO3 did not show much difference in the peak position and intensity, indicating that the main cationic skeleton of SJTU-COF was stable during microwave-assisted anion exchange. In the FT-IR spectrum of SJTU-COF-CF3SO3, the appearance of the characteristic C–F stretching band at 1030 cm-1 confirmed the inclusion of CF3SO3-.

|
Download:
|
| Fig. 2. (a) FT-IR spectra of SJTU-COFs. (b) XRD patterns of the SJTU-COFs. (c) N2 sorptionisotherm curves of SJTU-COFs. (d) Pore size distribution and pore volume profiles of SJTU-COFs. | |
{kind=link}
The PXRD patterns revealed that SJTU-COFs were crystalline materials. The most intense peak (2θ = 3.3°) and broad peak (27°) were assigned to the 100 and 001 facets. After anion exchange, the PXRD patterns (Fig. 2b) remain the same with that of SJTU-COF-Br. The peak positions in PXRD pattern of SJTU-COF-Br were the same with those of EB-COF:Br, which was prepared by a conventional heating method [25]. It indicated that the structure of SJTU-COF-Br was the same with that of EB-COF:Br. SJTU-COF-Br is a 2D layered hexagonal network. The π-π stacking distance was calculated to be 3.3 Å. After anion exchange, the PXRD patterns of SJTU-COF-Cl, SJTU-COF-AcO, and SJTU-COF-CF3SO3 remained the same with that of SJTU-COF-Br, indicating that crystal structure of cationic framework remained well. Thus, the main cationic skeletons of SJTU-COFs were stable during microwave-assisted anion exchange.
Scanning electron microscopy (SEM) images of SJTU-COFs showed the irregular shapes with aggregated submicrometer-size structures consisting of nano-size primary particles (Fig. S1 in Supporting information). In our future research, surface modification [36, 37] could be a good method for alleviating the aggregation. Transmission electron microscopy (TEM) images of SJTU-COFs revealed the nanoscale porous texture structure (Fig. S2 in Supporting information). To obtain quantitative information on the elemental structure of SJTU-COFs, C/H/N elemental analysis of SJTU-COFs was performed (Table S1 in Supporting information). In the case of SJTU-COF-Br, the mass fractions of C, H and N were found to be 64.03%, 4.23% and 8.01%, respectively. The mass fractions of C and N were a little bit higher than the theoretical value, which might result from the physically adsorbed water in SJTU-COF-Br. These experimental C/H/N elemental analysis values were close to theoretical values, indicating that SJTU-COF-Br had an elemental composition corresponding to the suggested structure. The experimental values of SJTU-COF-Cl, SJTU-COF-AcO and SJTU-COF-CF3SO3 were close to theoretical values of expectant structures, respectively. It indicated the complete exchange of Br- ions with expectant anions after microwave-assisted anion exchange, which could also be proved by the EDS elemental analysis (Fig. S3 in Supporting information). Taking SJTU-COF-Cl for example, the low signal of Br and the strong signal of Cl indicated that the Br- ions was completely exchanged by Cl- ion. Thermal gravimetric analysis (TGA) revealed that all the SJTU-COFs had great thermostability up to 450 ℃ (Fig. S4 in Supporting information). The cationic framework of SJTU-COFs exhibited great chemical stability of in hexane, THF, water, and 3 mol/L HCl (Fig. S5 in Supporting information).
The porosities of the SJTU-COFs were investigated by using nitrogen sorptionmeasurements at 77 K. The typical type I nitrogen sorption isotherms of SJTU-COFs (Fig. 2c) indicated microporosity with pore sizes around 1.6 nm (Fig. 2d) [38]. The Brunauer-Emmett-Teller (BET) surface areas of SJTU-COF-Br, SJTU-COF-AcO, SJTU-COF-Cl and SJTU-COF-CF3SO3 are calculated to be 944, 865, 954 and 879 m2/g, respectively (Table S2 in Supporting information). The BET surface areas of SJTU-COF-AcO and SJTU-COF-CF3SO3 are lower than that of SJTU-COF-Br and SJTU-COF-Cl, which can be explained by the increased size of AcO- and CF3SO3-.
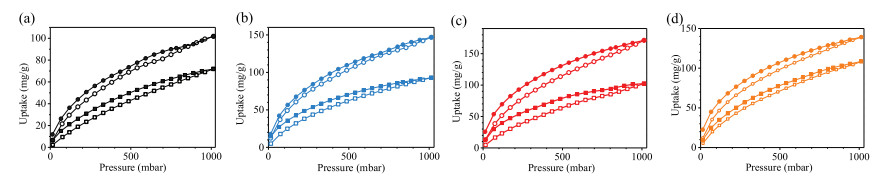
The CO2 sorption isotherms of SJTU-COFs were measured at 273 K and 298 K (Fig. 3). The SJTU-COF-Br uptakes CO2 with capacity of 101.9 m2/g at 273 K and 1 bar, which is superior to many COFs, including COF-5 (59 m2/g) [39], TDCOF-5 (92 m2/g) [40], and COF-103 (76 m2/g) [39] and ILCOF-1 (60 m2/g) [41]. The high capacity can be attributed to not only the abundant N—H sites on the pore walls interacting with polarizable CO2 molecules through hydrogen bond interactions [6], but also the cationic interface of the pore walls [42]. After ion exchange, the CO2 adsorption capacities of SJTU-COF-Br, SJTU-COF-Cl, SJTU-COF-AcO, and SJTU-COF-CF3SO3are found to be 101.9, 146.8, 171.2 and 139.1 m2/g at 273 K and 1 bar, respectively (Table S2). It is notable that CO2 capacity of SJTU-COF-AcO is the highest among these SJTU-COFs, which is increased to 1.7 times as compared with SJTU-COF-Br. To the best of our knowledge, the CO2 capacity of SJTU-COF-AcO is comparable to those of top-class members, including TpPa-1 (156 m2/g) [43], ACOF-1 (177 m2/g) [8] and carboxylic acid functionalized [HO2C]100%-H2P-COF (174 m2/g) [7] (Table S3 in Supporting information). The heat of adsorption (Qst) was calculated from the CO2adsorption data collected at different temperatures (273 and 298 K). At zero coverage, the heat of adsorption (Qst) of SJTU-COF-Br, SJTU-COF-Cl, SJTU-COF-AcO and SJTU-COF-CF3SO3 are found to be 29.1, 30.1, 34.2 and 25.3 kJ/mol (Fig. S6 and Table S2 in Supporting information). A possible explanation of the relatively high CO2uptake capacity of SJTU-COF-AcO might be related to the interaction between the carboxylate groups of the acetate anion with CO2. The CO2 molecules may interact with AcO- anion through a weak charge transfer interaction taking place between an oxygen atom of the COO- group of acetate anion (as a Lewis base) and the carbon atom of CO2 (acting as a Lewis acid). The pKb values of CF3SO3-, Br-, Cl- and AcO- anions are 28, 23, 21 and 9, respectively. The smaller the value of pKb, the stronger the base, the stronger the interaction between the anion and CO2, which is well consistent with the heat of adsorption results.

|
Download:
|
| Fig. 3. CO2 adsorption measurements for (a) SJTU-COF-Br, (b) SJTU-COF-AcO, (c) SJTU-COF-Cl, and (d) SJTU-COF-CF3SO3 at 273 K and 298 K. | |
{kind=link}
In summary, for the first time, a cationic SJTU-COF-Br was synthesized by a rapid microwave-assisted solvothermal method. Base on the SJTU-COF-Br, a set of SJTU-COFs that consist of the cationic framework coupled with different counter anionswere synthesized via channel-wall engineering strategy by microwave-assisted anion exchange. The resulting SJTU-COFs showed high crystallinity, stability and porosity. The heat of carbon dioxide adsorption was tuned by Lewis basicity of the counter ions. The stronger the base, the higher the heat of adsorption will be. The acetate anion containing COF showed a CO2 capacity of 171 mg/g at 273 K and 1 bar, which was increased to 1.7 times as compared with the pristine COF, and an isosteric enthalpy of adsorption of 34.2 kJ/mol. These results suggest that the anion exchange can be a novel strategy for channel-wall functional engineering, which provides a new way to tailor the pores of COFs for high-performance gas storage and separation.
AcknowledgmentsThis work is supported by the National Natural Science Foundation of China (Nos. 61774102, 81670958, 81873816, 61671299), the Shanghai Science and Technology Grant (No. 16JC1402000), Program for Shanghai Eastern Scholar and the Interdisciplinary Program of Shanghai Jiao Tong University (No. YG2016MS71). We also thank the Instrumental Analysis Center of Shanghai Jiao Tong University for the analysis support.
Appendix A. Supplementary dataSupplementary material related to this article canbefound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.05.012.
| [1] |
R.S. Haszeldine, Science 325 (2009) 1647-1652. DOI:10.1126/science.1172246 |
| [2] |
R. Dawson, D.J. Adams, A.I. Cooper, Chem. Sci. 2 (2011) 1173-1177. DOI:10.1039/c1sc00100k |
| [3] |
C. Chen, D.W. Park, W.S. Ahn, Appl. Surf. Sci. 292 (2014) 63-67. DOI:10.1016/j.apsusc.2013.11.064 |
| [4] |
R. Banerjee, A. Phan, B. Wang, et al., Science 319 (2008) 939-943. DOI:10.1126/science.1152516 |
| [5] |
X. Gong, Y. Wang, T. Kuang, ACS Sustainable Chem. Eng. 5 (2017) 11204-11214. DOI:10.1021/acssuschemeng.7b03613 |
| [6] |
H. Wei, S. Chai, N. Hu, et al., Chem. Commun. 51 (2015) 12178-12181. DOI:10.1039/C5CC04680G |
| [7] |
N. Huang, X. Chen, R. Krishna, D. Jiang, Angew. Chem. Int. Ed. 54 (2015) 2986-2990. DOI:10.1002/anie.201411262 |
| [8] |
Z. Li, X. Feng, Y. Zou, et al., Chem. Commun. 50 (2014) 13825-13828. DOI:10.1039/C4CC05665E |
| [9] |
S.Y. Ding, W. Wang, Chem. Soc. Rev. 42 (2013) 548-568. DOI:10.1039/C2CS35072F |
| [10] |
X. Feng, X. Ding, D. Jiang, Chem. Soc. Rev. 41 (2012) 6010-6022. DOI:10.1039/c2cs35157a |
| [11] |
X. Wang, X. Han, J. Zhang, et al., J. Am. Chem. Soc. 138 (2016) 12332-12335. DOI:10.1021/jacs.6b07714 |
| [12] |
S.Y. Ding, J. Gao, Q. Wang, et al., J. Am. Chem. Soc. 133 (2011) 19816-19822. DOI:10.1021/ja206846p |
| [13] |
Y. Wu, H. Xu, X. Chen, J. Gao, D. Jiang, Chem. Commun. 51 (2015) 10096-10098. DOI:10.1039/C5CC03457D |
| [14] |
H. Xu, J. Gao, D. Jiang, Nat. Chem. 7 (2015) 905-912. DOI:10.1038/nchem.2352 |
| [15] |
Y. Wu, D. Yan, Z. Zhang, M.M. Matsushita, K. Awaga, ACS Appl. Mater. Inter. 11 (2019) 7661-7665. DOI:10.1021/acsami.8b21696 |
| [16] |
Y. Wu, Z. Zhang, S. Bandow, K. Awaga, Bull. Chem.Soc. Jpn. 90 (2017) 1382-1387. DOI:10.1246/bcsj.20170247 |
| [17] |
C.R. DeBlase, K.E. Silberstein, T.T. Truong, H.D. Abruña, W.R. Dichtel, J. Am. Chem. Soc. 135 (2013) 16821-16824. DOI:10.1021/ja409421d |
| [18] |
Y. Han, N. Hu, S. Liu, et al., Nanotechnology 28 (2017) 33LT01. DOI:10.1088/1361-6528/aa7bb6 |
| [19] |
C.R. Mulzer, L. Shen, R.P. Bisbey, ACS Cent. Sci. 2 (2016) 667-673. DOI:10.1021/acscentsci.6b00220 |
| [20] |
X. Zhang, Z. Wang, L. Yao, et al., Mater. Lett. 213 (2018) 143-147. DOI:10.1016/j.matlet.2017.11.002 |
| [21] |
X. Zhang, L. Yao, S. Liu, et al., Mater. Today Energy 7 (2018) 141-148. DOI:10.1016/j.mtener.2018.01.003 |
| [22] |
S. Liu, L. Yao, Y. Lu, et al., Mater. Lett. 236 (2019) 354-357. DOI:10.1016/j.matlet.2018.10.131 |
| [23] |
L. Li, S. Liu, Q. Zhang, et al., Acta Phys. Chim. Sin. 33 (2017) 1960-1977. |
| [24] |
L. Wu, J. Zheng, L. Wang, et al., Angew. Chem. Int. Ed. 58 (2019) 811-815. DOI:10.1002/anie.201811784 |
| [25] |
H. Ma, B. Liu, B. Li, et al., J. Am. Chem. Soc. 138 (2016) 5897-5903. DOI:10.1021/jacs.5b13490 |
| [26] |
N. Huang, R. Krishna, D. Jiang, J. Am. Chem. Soc. 137 (2015) 7079-7082. DOI:10.1021/jacs.5b04300 |
| [27] |
S. Chai, H. Liu, X. Zhang, et al., Appl. Surf. Sci. 384 (2016) 539-543. DOI:10.1016/j.apsusc.2016.05.068 |
| [28] |
P.L. Cheung, S.K. Lee, C.P. Kubiak, Chem. Mater. 31 (2019) 1908-1919. DOI:10.1021/acs.chemmater.8b04370 |
| [29] |
C.L. Yao, J.C. Li, W. Gao, et al., Chem. Eur. J. 24 (2018) 11051-11058. DOI:10.1002/chem.201800363 |
| [30] |
K. Duan, J. Wang, Y. Zhang, et al., J. Membr. Sci. 572 (2019) 588-595. DOI:10.1016/j.memsci.2018.11.054 |
| [31] |
K. Kamiya, Impact 2018 (2018) 57-59. DOI:10.3390/pharmaceutics10020057 |
| [32] |
M. Wu, Y. Yang, Chin. Chem. Lett. 28 (2017) 1135-1143. DOI:10.1016/j.cclet.2017.03.026 |
| [33] |
Y. Han, Q. Zhang, N. Hu, et al., Chin. Chem. Lett. 28 (2017) 2269-2273. DOI:10.1016/j.cclet.2017.10.024 |
| [34] |
H. Dong, D. Zhang, R. Fang, et al., Synthetic Commun 48 (2018) 1227-1234. DOI:10.1080/00397911.2018.1440316 |
| [35] |
D.D. Zhang, Y.L. Liu, Y. Wang, et al., Chin. Chem. Lett. 27 (2016) 563-565. DOI:10.1016/j.cclet.2016.01.047 |
| [36] |
Y. Wang, X. Gong, Adv. Mater. Interfaces 4 (2017) 1700190. DOI:10.1002/admi.201700190 |
| [37] |
Y. Wang, X. Gong, J. Mater. Chem. A 5 (2017) 3759-3773. DOI:10.1039/C6TA10474F |
| [38] |
L. Wei, W. Lu, H. Wei, C. Chen, Z. Hou, Micropor. Mesopor. Mat. 241 (2017) 58-65. DOI:10.1016/j.micromeso.2016.12.003 |
| [39] |
H. Furukawa, O.M. Yaghi, J. Am. Chem. Soc. 131 (2009) 8875-8883. DOI:10.1021/ja9015765 |
| [40] |
Z. Kahveci, T. Islamoglu, G.A. Shar, R. Ding, El-Kaderi H.M., Cryst. Eng. Comm. 15 (2013) 1524-1527. DOI:10.1039/C2CE26487K |
| [41] |
M.G. Rabbani, A.K. Sekizkardes, Z. Kahveci, et al., Chem.-Eur. J. 19 (2013) 3324-3328. DOI:10.1002/chem.201203753 |
| [42] |
N. Huang, P. Wang, M.A. Addicoat, T. Heine, D. Jiang, Angew. Chem. Int. Ed. 41 (2017) 6010-6016. DOI:10.1002/anie.201611542 |
| [43] |
S. Kandambeth, A. Mallick, B. Lukose, et al., J. Am. Chem. Soc. 134 (2012) 19524-19527. DOI:10.1021/ja308278w |