 2020, Vol. 31
2020, Vol. 31 


Chemical modification of proteins has emerged as an invaluable tool for creating engineered proteins [1] and for the study of protein functions and behaviour in living system [2, 3]. For example, protein chemically modified with fluorophore shows great potential in biomaterial [4], and biosensors [5, 6]. In pharmaceutical applications, protein modified with polyethylene glycol (PEG) is widely used for extending the protein half-time and reducing immunogenicity [7]. Antibody conjugated with cytotoxic compound, known as antibody drug conjugate (ADC) [8], has shown great potential in cancer therapy. In most cases, it is critical to precisely control the site of modification. Especially for therapeutic proteins, homogeneous modification is a key factor to be considered for quality and safety control [9]. To this end, a wide variety of methods have been developed for protein modification.
The most frequently used method to modify proteins is through naturally occurring nucleophilic or reactive amino acid such as lysine, cysteine, tryptophan [10]. Cysteine is unique among the 20 canonical amino acids due to its low abundance and the thiol group on the side chain has robust nucleophilic ability [11], which can be used for fast and convenient protein chemical conjugation under mild conditions [12]. Therefore, engineering an unpaired cysteine is one of the most commonly used approaches for protein site-specific labelling. Over the years, a great number of thiol reactive reagents have been developed and commercially available, providing a cheap and convenient way for protein site selective conjugation. The primary thiol alkylation reagents [13] include maleimides, iodoacetamides, benzylic halides and bromomethylketones, which react with thiols to generate stable thioether products. In particular, the cysteine-maleimide reaction is among the most commonly used reactions for protein modification, and is widely used for the detection of conformational changes, assembly of multi-unit complexes [14, 15] and ligand-binding processes [16]. Over the past decade, a number of bio-orthogonal reactive groups has been developed for peptide [17] and protein site-specific labelling, including copper-catalyzed azide-alkyne cycloaddition (CuAAC), strain-promoted azide-alkyne cycloaddition (SPAAC), and inverse-electron-demand Diels-Alder (IEDDA), etc. [18]. However, the large-scale synthesis and storage for many of these reaction handles is still difficult and expensive. While cysteine reactive reagents are easy to obtain and therefore are still among the most popular choices for protein site-specific labelling and detection.
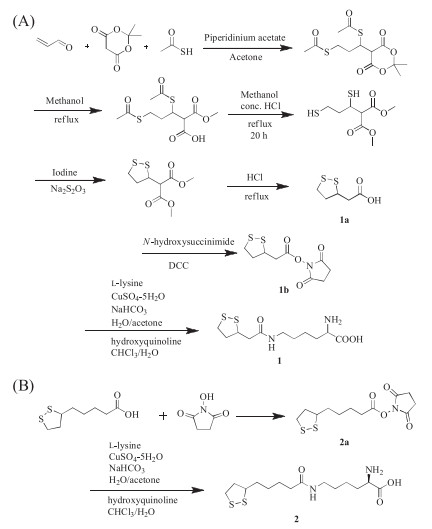
However, for proteins containing internal disulfide bonds, it is technically challenging to introduce an additional unpaired single cysteine in the recombinant expression these proteins, which usually leads to protein misfolding and oligomerization. The resulted proteins also suffer from significantly decreased yield and activities [19], and often require complicated purification processes [20]. This is more problematic for therapeutically important proteins, since most of them contain internal disulfide bonds and require high yield for production [21]. To address this issue, several methods have been established for single cysteine engineering, which typically involves protein refolding or removal of active cysteines [22]. We wonder if it is possible to introduce two thiols simultaneously, which can form internal disulfides with each other, therefore would not interfere with the native cysteines on the protein of interest. To achieve this goal, two unnatural amino acids (UAA), trisnorlipoyllysine (Tsn-K) (1) and lipoyllsine (Lip-K) (2) containing self-paired dithols were designed and synthesized. The synthesis of these two compounds was shown in Scheme 1. The total yield was 8% for 1 and 30% for 2 through eight and three steps' reactions, respectively. Their identities were confirmed by ESI-MS, 1H NMR, and 13C NMR.

|
Download:
|
| Scheme 1. Synthesis of trisnorlipoyllysine (Tsn-K, 1) (A) and lipoyllsine (Lip-K, 2) (B). | |
{kind=link}
To site-specifically introduce these two amino acids into proteins, we applied a non-canonical amino acid mutagenesis strategy using an expanded genetic code [23]. The pyrrolysyl-tRNA synthetase (PylRS)/tRNACUA pair have been widely used for genetic incorporation of many pyrrolysine analogues. Based on the crystal structure of M. maize PylRS pyrrolysyl-AMP (PDB code 2ZIM), we constructed a library with four positions (L270, Y271, L274, C313) randomly mutated to NNK (N = any nucleotide, K = G or T) and a fixed mutation on Y349F (Fig. 1A), which has been previously reported to increase aminoacylation efficiency [24]. The library was then subjected to positive-negative-positive selection to identify PylRS variants that can charge these two UAAs into pyrrolysyl tRNA according to previously reported method [25]. Finally two consistent sequence were obtained and named LKRS-1 and LKRS-2 (LKRS-1:Y271A/Y349F and LKRS-2:Y271 G/Y349 F) for following characterizations.

|
Download:
|
| Fig. 1. (A) X-ray crystal structure of the MmPylRS complex with pyrrolysyl-AMP. The structure is from PDB entry 2ZIM. (B) UAAs incorporation with different aaRSs. (C) GFP expressed in the presence and absence of 1 mmol/L UAAs with LKRS-1. (D) Incorporation of Lip-K and Tsn-K into EGFP-Y39TAG expressed in mammalian cells. | |
{kind=link}
To evaluate the incorporation efficacy of these UAAs with evolved aaRSs, we cloned the aaRS genes into pUltra plasmid [26], and co-transformed with pET22B-T5-sfGFP-Y151TAG into BL21(DE3) cell. The suppression efficiency was measured with the intensity of GFP fluorescence. As shown in Fig. 1B, a significant fluorescence can be observed in the presence of 1 mmol/L UAAs. Among three aaRSs tested, including the wildtype PylRS, LKRS-1 showed the best activity for both Tsn-K and Lip-K incorporation, and was therefore selected for the following studies.
To further validate the incorporation of these two UAAs in GFP, the GFP mutants containing amber stop codon were expressed in the presence or absence of 1 mmol/L UAA. The resulting proteins were purified by Ni-NTA column and characterized by SDS-PAGE analysis. As shown in Fig. 1C, the full-length GFP can only be observed in the presence of 1 mmol/L UAA. The identification of these UAAs was confirmed by high-resolution mass spectrometry [quadrupole-time-of-flight/MS (QTOF/MS)]. The mass spectra trace revealed that Lip-K GFP and wild type GFP give an observed mass of 27, 750 Da and 27, 597 Da, respectively. The mass difference of 153 Da agrees with the molecular difference between tyrosine and Lip-K, which confirmed the site-specific incorporation of Lip-K into GFP (Fig. S1 in Supporting information). On the other hand, the Tsn-K GFP shows observe mass of 26, 778 Da, which matches with the incorporation of Tsn-K (Fig. S2 in Supporting information). However, synthesis of Tsn-K is practically more difficult than Lip-K, so Lip-K was the priority used in the following experiment because of its high efficiency and low cost.
As previously reported, the pyrrolysyl-tRNA synthetase/tRNAPyl pair has been shown to be orthogonal in mammalian cells [11, 27]. To test the feasibility of incorporating these two UAAs in mammalian cells, a plasmid encoding LKRS-1 under the control of CMV promoter, as well as a U6 promoter-drived tRNAPyl cassette was constructed. Incorporation of the UAAs were evaluated by co-transfection with PCMV-EGFP (Y39TAG) (Fig. 1D). The suppression efficiency was evaluated by the brightness of green fluorescence in the presence of 1 mmol/L UAAs. Agreeing with the bacterial result incorporation results, the Lip-K showed a significantly enhanced fluorescence in mammalian cell and was carried out for further studies.
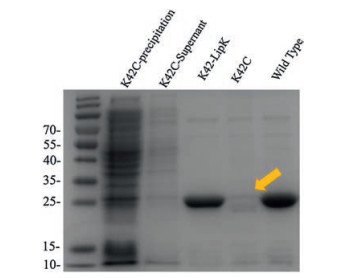
Disulfide containing proteins can be recombinantly expressed in the periplasmic space of E. coli to afford soluble protein without refolding [28]. However, it is generally non-compatible with engineered single cystine. To investigate whether the self-paired disulfide bond would solve this problem, we recombinantly expressed Herceptin single-chain variable fragment (scFv) containing a natural pair of disulfide. Plasmid encoding Herceptin scFv were periplasmic expressed in BL21 (DE3) and purified by Ni-NTA. The yield of wild type protein was 5 mg/L (Fig. S3 in Supporting information). For UAA incorporation, six different amber codon mutations were generated. The proteins were expressed with 1 mmol/L Lip-K, and purified similarly to the wildtype protein. SDS-Page analysis showed that E1 and K42 have the best expression yield comparing to wild type proteins (Fig. S4 in Supporting information). As a comparison, K42C mutation, which contains a single unpaired cysteine, could not afford soluble protein, while K42-LipK showed no influence on protein expression (Fig. 2).

|
Download:
|
| Fig. 2. SDS-PAGE analysis of periplasmic expressed Herceptin scFv mutants. K42C scFv mutant has a very low expression level (lanes 1 and 2) and the purified protein cannot be obtained by Ni column purification (lane 4). While under the same condition, purified protein can be obtained for wild type scFv (lane 5) and K42LipK mutant (lane 3) with comparable yield. | |
{kind=link}
Next, we examined whether this self-paired disulfide bond can be selectively reduced without disturbing the internal disulfide bond. To allow selective chemical conjugation, Lip-K was introduced on the surface of the protein, whereas the natural disulfide bonds typically are buried in the protein interiorly. A mild reducing reagent tris(2-carboxyethyl)phosphine (TCEP) was applied at 1 mmol/L concentration to preferentially reduce engineered, exposed disulfide bonds. As shown in Fig. 3A, high-resolution QTOF/MS analysis revealed that the reduced protein shows an observable +2 mass compared with wild type protein, which is consistent with the reduction of the one pair of disulfide bond. To further confirm this result, the reduced sample were treated with iodoacetamide (IAM), an alkylating agent used for free thiol labelling [13]. Indeed, QTOF/MS trace revealed a 114 Da addition, which was resulted from the reaction of thiol with two equivalents IAM. As a comparison, no mass difference was observed in the control group, indicating that the modification was indeed happened on the reduced thiols.

|
Download:
|
| Fig. 3. (A) Mass spectral analysis of purified scFv-K42-LipK protein under four different treatment conditions. (B) FACS analysis of FITC random labeled scFv and iFluor647 site-specifically labeled scFv treated SK-BR-3 cells. | |
{kind=link}
To study whether this UAA mutagenesis and chemical conjugation will influence protein activity, the function of Herceptin scFv was examined by measuring its binding to Her2-expressing (binding target of Herceptin) cancer cells. The fluorophore was first site-specifically conjugated to the introduced dithiol through chemical conjugation. Briefly, reduced scFv-LipK was treated with 10 mmol/L maleimide-iFluorTM647. As a contrary, wild type scFv was randomly labelled with FITC through lysine conjugation. The conjugated antibodies were tested on Her2-expressing breast cancer cells, SK-BR-3. As shown in Fig. 3B, FACS analysis indicated that iFluorTM647 conjugated scFv-LipK retained binding ability to SK-BR-3 cells, suggesting that the designed LipK can be used for site-specific protein conjugation using thiol labelling reagents without influencing protein activity.
In conclusion, we have established a new method for introducing thiols into proteins containing natural disulfide bond. The introduction of dithiol containing amino acid has no influence on protein activity and do not require protein refolding. This method affords two thiols site-specially into protein of interest, which allows chemical modifications with cheap and convenient cysteine labelling probes. This is especially important in the production of chemically modified therapeutic proteins, which often contains natural disulfide bonds and requires economical reagents for batch production. In addition, this method should be compatible with other bio-orthogonal protein modification approaches, which could afford multiple site-specific conjugations. The ability to introduce di-thiol provides insights into protein modification and has profound influence on production of biotherapeutics.
AcknowledgmentsThis work was financially supported by National Key Research and Development Program of China (No. 2016YFA0201400), the National Natural Science Foundation of China (No. 21778005), Peking University Health Science Center (Nos. BMU20160537 and BMU2017QQ006). T. Liu thanks the Youth Thousand-Talents Program of China for support.
Appendix A. Supplementary dataSupplementary material related to this article canbefound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.04.075.
| [1] |
S. Leng, Q.L. Qiao, Y. Gao, et al., Chin. Chem. Lett. 28 (2017) 1911-1915. DOI:10.1016/j.cclet.2017.03.034 |
| [2] |
S. Liébana, G.A. Drago, Essays Biochem. 60 (2016) 59-68. DOI:10.1042/EBC20150007 |
| [3] |
C.D. Spicer, B.G. Davis, Nat. Commun. 5 (2014) 4740. DOI:10.1038/ncomms5740 |
| [4] |
D.P. Nair, M. Podgórski, S. Chatani, et al., Chem. Mater. 26 (2014) 724-744. DOI:10.1021/cm402180t |
| [5] |
S. Sakamoto, I. Hamachi, Anal. Sci. 35 (2019) 5-27. DOI:10.2116/analsci.18R003 |
| [6] |
Y. Yang, H. Wang, Y.L. Wei, et al., Chin. Chem. Lett. 28 (2017) 2023-2026. DOI:10.1016/j.cclet.2017.08.051 |
| [7] |
P. Bailon, C.Y. Won, Expert Opin. Drug Deliv. 6 (2009) 1-16. DOI:10.1517/17425240802650568 |
| [8] |
A. Beck, L. Goetsch, C. Dumontet, N. Corvaïa, Nat. Rev. Drug Discov. 16 (2017) 315-337. DOI:10.1038/nrd.2016.268 |
| [9] |
P. Akkapeddi, S.A. Azizi, A.M. Freedy, et al., Chem. Sci. 7 (2016) 2954-2963. DOI:10.1039/C6SC00170J |
| [10] |
P. Wang, S. Zhang, Q. Meng, et al., Org. Lett. 17 (2015) 1361-1364. DOI:10.1021/acs.orglett.5b00005 |
| [11] |
T. Liu, Y. Wang, X. Luo, et al., Proc. Natl. Acad. Sci. U. S. A. 113 (2016) 5910-5915. DOI:10.1073/pnas.1605363113 |
| [12] |
J.M. Chalker, G.J.L. Bernardes, Y.A. Lin, B.G. Davis, Chem. Asian J. 4 (2009) 630-640. DOI:10.1002/asia.200800427 |
| [13] |
S. Sechi, B.T. Chait, Anal. Chem. 70 (1998) 5150-5158. DOI:10.1021/ac9806005 |
| [14] |
A. Sánchez, E. Pedroso, A. Grandas, Chem. Commun. 49 (2013) 309-311. DOI:10.1039/C2CC35357A |
| [15] |
B.T. Houseman, E.S. Gawalt, M. Mrksich, Langmuir 19 (2003) 1522-1531. DOI:10.1021/la0262304 |
| [16] |
J.M.J.M. Ravasco, H. Faustino, A. Trindade, P.M.P. Gois, Chem. Eur. J. 25 (2019) 43-59. DOI:10.1002/chem.201803174 |
| [17] |
Y.J. Pu, H. Yuan, M. Yang, B. He, Z.W. Gu, Chin. Chem. Lett. 24 (2013) 917-920. DOI:10.1016/j.cclet.2013.06.015 |
| [18] |
B.L. Oliveira, Z. Guo, G.J.L. Bernardes, Chem. Soc. Rev. 46 (2017) 4895-4950. DOI:10.1039/C7CS00184C |
| [19] |
A. Schmiedl, F. Breitling, C.H. Winter, I. Queitsch, S. Dübel, J. Immunol. Methods 242 (2000) 101-114. DOI:10.1016/S0022-1759(00)00243-X |
| [20] |
M. Moghadam, A. Ganji, A. Varasteh, R. Falak, M. Sankian, Rep. Biochem. Mol. Biol. 4 (2015) 19-24. |
| [21] |
C.A. Thomson, M. Olson, L.M. Jackson, J.W. Schrader, PLoS One 7 (2012) E49891. DOI:10.1371/journal.pone.0049891 |
| [22] |
I. Sengupta, J.B. Udgaonkar, Protein Expr. Purif. 140 (2017) 1-7. DOI:10.1016/j.pep.2017.07.014 |
| [23] |
L. Wang, A. Brock, B. Herberich, P.G. Schultz, Science 292 (2001) 498-500. DOI:10.1126/science.1060077 |
| [24] |
T. Yanagisawa, R. Ishii, R. Fukunaga, et al., Chem. Biol. 15 (2008) 1187-1197. DOI:10.1016/j.chembiol.2008.10.004 |
| [25] |
H. Neumann, S.Y. Peak-Chew, J.W. Chin, Nat. Chem. Biol. 4 (2008) 232-234. DOI:10.1038/nchembio.73 |
| [26] |
A. Chatterjee, S.B. Sun, J.L. Furman, H. Xiao, P.G. Schultz, Biochemistry 52 (2013). DOI:10.1021/bi4000244 |
| [27] |
H. Xiao, W. Xuan, S. Shao, T. Liu, P.G. Schultz, ACS Chem. Biol. 10 (2015) 1599-1603. DOI:10.1021/cb501055h |
| [28] |
R. Rouet, D. Lowe, K. Dudgeon, et al., Nat. Protoc. 7 (2012) 364-373. DOI:10.1038/nprot.2011.448 |