 2020, Vol. 31
2020, Vol. 31 


b University of Chinese Academy of Sciences, Beijing 100049, China
MicroRNAs (miRNAs) are a group of small non-coding RNA molecules (containing about 22 nucleotides) that were widely found in plants, animals, and some viruses [1-6]. Studies have shown that miRNAs play important roles in RNA silencing, post-transcriptional regulation of gene expression [7-10]. Most recently, miRNAs were found expression change in several pathological diseases or solid tumors [11-13]. Thus the indicator of miRNAs can be used as a tool for cancer diagnosis and prognosis judgment [11, [14-18]. Recently, a variety of miRNA detection methods based on nucleic acid amplification technology have been developed, such as reverse transcription-polymerase chain reaction (RT-PCR) [19, 20], rolling-circle amplification (RCA) [21-23], exponential amplification reaction (EXPAR) [24-28], loop-mediated isothermal amplification (LAMP) [29], ligation-LAMP [30], target-triggered LAMP (TT-LAMP) [31], RCA-LAMP [32], along with traditional approaches like northern blotting analysis [33], microarray [34] etc. Those nucleic acid detection technologies have amplified the target RNAs to a significant amount with a low detection limit, but still have some shortcomings. For example, RT-PCR represents one of the most widely used detection methods so far [19]. Since the sequence of miRNA is only about 20 bases, it is generally necessary to use a loop primer or linear primer to complete reverse transcriptase reactions and obtain cDNA. A following real-time polymerase chain reaction (qPCR) with earlier obtained cDNA amplified the sequence and facilitated the precise measurement of the expression level of those miRNAs, which usually need about 3–4 h. Nonetheless, these methods based on isothermal amplification usually required multiple steps [27, 28, 30] which was time-consuming [27] and may cause false positive signals when triggered by LAMP product [28]. Other methods, such as northern blotting analysis or microarray, were always accomplished with weak and non-specific signals [33, 34]. Based on these insufficiencies, improved strategies based on isothermal amplification, such as LAMP, were continuously developed by using miRNA as an outer primer probe [29, 31]. Considerable progress has been made in this area and significant detection limits were obtained [32], for example, Prof. Li and his co-workers initiated the use of isothermal amplification-based technologies for miRNA detection and significant results were obtained in several cases [26, 29, 31, 32]. Despite of these elegant examples [28, 32], highly selective and sensitive for miRNA detection methods are always required to fulfil current diagnosis requirements.
Isothermal amplification such as LAMP has attracted extensive attention in the last decade as an alternative to PCR [35-38]. The key difference between the two is that LAMP does not need thermal cycling, and the polymerase can amplify the target sequence at a facile temperature, 60 - 65 ℃, for example [39]. That could be easily achieved by water baths or heating modules, thus eliminating the need for intricate instruments [35]. The high sensitivity of the LAMP lies in the ability to form dumb-bell structure, and it can be cyclically amplified with the combination of inner primers and loop primers [35-38]. Therefore, LAMP has been recognized as a simple and time-saving method for on-site detection of DNAs [40, 41]. However, by using miRNA as the outer primer probe, by-products were obtained without triggering the cyclic amplification process, which may cause a relatively weak sensitivity [31]. With our continuing interests in the selective recognition and modulation of RNA [42-46], herein, we disclose a sophisticated design by combining asymmetric PCR and LAMP (AP-LAMP) for an ultrasensitive detection of miRNAs with high sensitivity (10 amol/L) and selectivity. Considering the inner primer and loop primer both participated in the cyclic amplification process and inner primer was too long to be replaced by miRNA, we replaced miRNA with an equivalent length loop primer and achieved excellent amplification comparing with that without loop primer. The detection process can be accomplished in a short time (~90 min), and by using SYBR dye for the amplification detection this may serve as a reliable choice for an on-site ultrasensitive detection of miRNA.
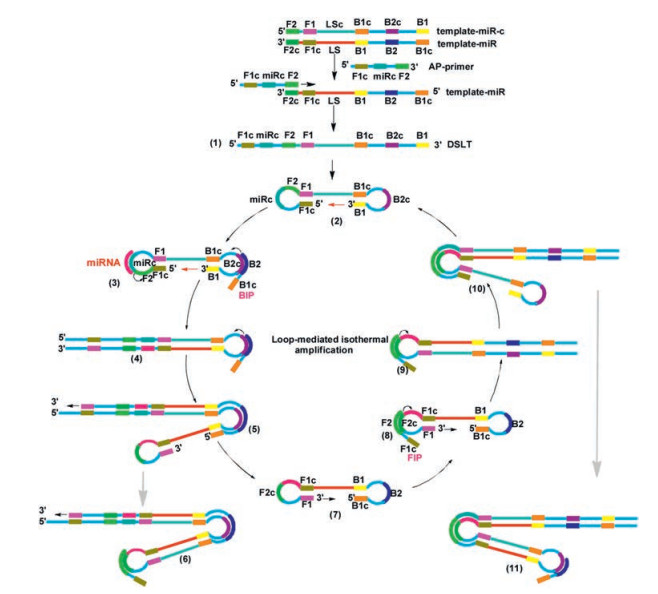
Our approach for the development of a sequence selective and detection system is depicted in Scheme 1. This method relied on asymmetric PCR to generate an extended ssDNA strand template, which could not be obtained by commercial sources. In the initial step of asymmetric PCR, two primer extension templates were used, one of which (primer extension template, template-miR) was used for strand replacement DNA synthesis in the asymmetric PCR process. The sequences of template-miR and template-miR-c were designed as follows. The template-miR contained F2c (complementary to F2), F1c (complementary to F1), linking strand (LS), B1, B2 and B1c (complementary to B1). The template-miR-c contained F2, F1, LS-c (complementary to LS), B1c (complementary to B1), B2c (complementary to B2) and B1 (Table 1). An asymmetric PCR primer (AP-primer, typically 60 - 70 nt) was designed which contained F1c, miRc (miRNA complementary) and F2. The AP-primer hybridized to F2c in the template-miR and initiated a complementary stand elongation to afford an extended ssDNA (typically 140 - 160 nt), while served as a double stem-loop template (DSLT, structure 1) through the production of a dumb-bell form structure 2 upon heating at 60 - 65 ℃ for the LAMP amplification reaction.

|
Download:
|
| Scheme 1. Detailed reaction mechanism of AP-LAMP for miRNA detection. | |
|
|
Table 1 Sequences of oligonucleotides used in this study.a |

{kind=link}
The design and application of asymmetric PCR step and the subsequent LAMP amplification ensured high specificity of the target miRNA sequence recognition. Moreover, fluorescent dye SYBR Green Ⅱ was added into the LAMP reaction system. In the presence of Bst 2.0 WarmStart DNA polymerase and dNTPs, the fluorescent dye inserted into the amplification product during the process, producing a strong fluorescent signal through real-time PCR detection. Thus an ultrasensitive amplification was achieved in a short time. Generally the time required for the amplification by adding specific loop primer is 1/3 to 1/2 of that without loop primer [38].
In order to demonstrate the efficiency, the specificity and the ease of this detection method, we chose miRNA-34 (miR-34) as the model target and screened those sequences that met LAMP requirements (Table 1). Since the structure 1 was critical for the efficiency of LAMP, the sequences of the primers were optimized so that their melting temperatures (Tm) fell within certain ranges. The Tm value of F1/F1c was set slightly higher than that of miRNA/miRNA-c for the formation of dumb-bell DNA. DNA polymerase was another important factor for this process. The best amplification was obtained with Bst 2.0 WarmStart DNA polymerase. Key factors in the LAMP reaction such as different fluorescent dyes, reaction temperature, amount of Bst 2.0 WarmStart DNA polymerase and the amount of DSLT were carefully optimized. As shown in Figs. S3-S6 (Supporting information), we have carefully screened the optimal dye, the impact of different temperature, DNA polymerase and the impact on the result of DSLT amount. Thus the reaction was carried at 65 ℃ for 90 min and the products of asymmetric PCR were confirmed by gel electrophoresis as shown in Fig. S1 (Supporting information) and LAMP result were confirmed by PAGE result as shown in Fig. S7 (Supporting information).
LAMP is highly sensitive and able to detect as few as several copies of nucleic acidsin the reaction mixture. Thus we used miR-34a to investigate the sensitivity of the AP-LAMP technique. As shown in Fig. 1, in a 90 min LAMP reaction, 10 amol/L of miR-34 was amplified to a detectable level, and the sensitivity is 10 times higher than previously report [31].

|
Download:
|
| Fig. 1. (a) The real-time fluorescence curves of the LAMP system produced at different concentrations of miR-34a from 10 amol/L to 100 fmol/L. The negative control (random sequence) was treated in the same way as the target miRNA but without adding miR-34a. (b) The relationship between the T values and the logarithm (lg) of the concentrations (mol/L) of miR-34a. Error bars represent the standard deviation of three independent experiments. | |
{kind=link}
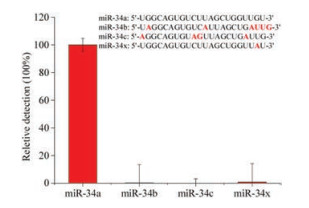
To further validate the established method, we used three members of the miRNA-34a family with single to six-base mutations, namely miR-34b, miR-34c and miR-34x, and the detail information were shown in Table 1. The relative detection efficiency of miR-34a was designated as 100%, and the relative detection efficiency of miR-34b, miR-34c and miR-34x were determined and calculated as 0.168%, 0.006% and 0.875%, respectively. The relative detection result was shown in Fig. 2 and the detail calculation information was shown in Fig. S2 (Supporting information). Thus we have confirmed that the established method has excellent specificity and can meet the requirement of single base variation detection.

|
Download:
|
| Fig. 2. The relative detection of the miR-34a miRNA family members and single base change oligo (miR-34x) with the AP-LAMP method. In the sequence list of miR-34a miRNA family members and miR-34x, the different bases compared to miR-34a in other miRNAs are marked in red. | |
{kind=link}
In order to verify that the established method can be used for the detection of other miRNAs, we randomly chose miR-155 to evaluate the method, then replaced the miRNAc-34 in AP-primer to miRNAc-155 for the measurement of miR-155. By using miR-34a as a negative control, a concentration as low as 10 amol/L of miR-155 was detectable, and the result was shown in Fig. 3. The data demonstrating that this method is simple and easy to perform once the appropriate AP-primers were prepared. To evaluate the specificity of established method, other three types of miRNA were detected, and the result was shown in Fig. S8 (Supporting information). The result confirmed that the established AP-LAMP method has highly specificity for miRNA detection.

|
Download:
|
| Fig. 3. (a) The real-time fluorescence curves of the LAMP system produced at different concentrations of miR-155 from 10 amol/L to 100 fmol/L. The negative control miR-34a was treated in the same way as the target miRNA but without adding miR-155. (b) The relationship between the T values and the logarithm (lg) of the concentrations (mol/L) of miR-155. Error bars represent the standard deviation of three independent experiments. | |
{kind=link}
miRNAs may regulate the expression of various oncogenes or tumor suppressor genes, thus some miRNAs were used as biomarkers to evaluate the tumor initiation, progression and response to treatment in cancer patients [47], [48], [49]. It was reported that miR-34a functions as tumor suppressors and inhibits the migration of human breast cancer cells through down-regulation of the targets (including Bcl-2 and SIRT1) [47], [48], [49]. Thus the detection of miR-34a is a useful biomarker in diagnosis of human breast cancer [11, 50]. To further demonstrate the utility of the assay, we examined the expression of miR-34a in total RNA of human MCF7 breast cancer cells (MCF7) from clinical samples. As shown in Fig. 4 and Fig. S9 (Supporting information), the relative expression of miR-34a in MCF7 human breast cancer cells was calculated as 0.46 ± 0.13, which was significantly lower than that of normal 293T cell. To further confirm this, we used the standard method of RT-qPCR to verify the data, and as shown in Fig. S10 (Supporting information) confirmed that the amount of miR-34a in MCF7 human breast cancer cells was around 0.57 ± 0.13 compared to 293 T cell. Thus it was confirmed that the expression of miR-34a in human breast cancer cells was lower than that of normal cell and current established method is capable of detecting the amount of specific miRNA in clinic samples.

|
Download:
|
| Fig. 4. The expression between the MCF7 breast cancer cell and normal 293T cell with the same amounts of total RNA input. Error bars represent the standard deviation of three independent experiments, *P ≤ 0.05. | |
{kind=link}
In conclusion, we have developed a novel method by combining asymmetric PCR and LAMP which amplifies specific miRNA with high specificity, efficiency and rapidity under isothermal conditions. This method employed several designed primer probes to recognize the target sequence with high selectivity. To the best of our knowledge, this is the first example of using miRNA as a loop primer probe in LAMP reaction. Compared with the traditional miRNA detection method such as RT-qPCR, this approach usually completed within 90 min, which provides a reliable choice for on-site ultrasensitive and rapid detection of miRNA. Besides, the excellent detection limit and sequence specificity will definitely ensure it functions as a valuable tool in various life science fields, clinical medicines and diagnosis of infectious diseases, for example. These advances will be reported in due courses.
AcknowledgmentsThis work was supported by the National Key R & D Program of China (Nos. 2017YFA0208100, 2016YFA0602900), National Natural Science Foundation of China (Nos. 91853124, 21778057 and 21420102003) and Chinese Academy of Sciences.
Appendix A. Supplementary dataSupplementary material related to this article can befound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.05.003.
| [1] |
R. Sunkar, Y.F. Li, G. Jagadeeswaran, Trends Plant Sci. 17 (2012) 196-203. DOI:10.1016/j.tplants.2012.01.010 |
| [2] |
K. Rogers, X. Chen, Plant Cell 25 (2013) 2383-2399. DOI:10.1105/tpc.113.113159 |
| [3] |
J.C. Carrington, V. Ambros, Science 301 (2003) 336-338. DOI:10.1126/science.1085242 |
| [4] |
Y. Moran, M. Agron, D. Praher, U. Technau, Nat. Ecol. Evol. 1 (2017) 27. DOI:10.1038/s41559-016-0027 |
| [5] |
R. Triboulet, B. Mari, Y.L. Lin, et al., Science 315 (2007) 1579-1582. DOI:10.1126/science.1136319 |
| [6] |
C.L. Jopling, M. Yi, A.M. Lancaster, S.M. Lemon, P. Sarnow, Science 309 (2005) 1577-1581. DOI:10.1126/science.1113329 |
| [7] |
S. Jonas, E. Izaurralde, Nat. Rev. Genet. 16 (2015) 421-433. DOI:10.1038/nrg3965 |
| [8] |
X. Wang, M. Li, Z. Wang, et al., J. Biol. Chem. 290 (2015) 3925-3935. DOI:10.1074/jbc.M114.596866 |
| [9] |
A. Molnár, F. Schwach, D.J. Studholme, E.C. Thuenemann, D.C. Baulcombe, Nature 447 (2007) 1126-1129. DOI:10.1038/nature05903 |
| [10] |
X. Yin, J. Hu, H. Xu, Chin. Chem. Lett. 29 (2018) 1029-1032. DOI:10.1016/j.cclet.2018.04.027 |
| [11] |
K.B. Reddy, Cancer Cell Int. 15 (2015) 38. DOI:10.1186/s12935-015-0185-1 |
| [12] |
Y. Liu, Q. Cai, P.P. Bao, et al., Breast Cancer Res. Treat. 152 (2015) 183-191. DOI:10.1007/s10549-015-3460-x |
| [13] |
A. Bakkar, M. Alshalalfa, L.F. Petersen, et al., Mol. Biol. Rep. 43 (2016) 229-240. DOI:10.1007/s11033-016-3948-4 |
| [14] |
M.V. Iorio, P. Casalini, E. Tagliabue, S. Menard, C.M. Croce, Eur. J. Cancer 44 (2008) 2753-2759. DOI:10.1016/j.ejca.2008.09.037 |
| [15] |
D. Wu, X. Xie, A.A. Kadi, Y. Zhang, Chin. Chem. Lett. 29 (2018) 1098-1104. DOI:10.1016/j.cclet.2018.04.030 |
| [16] |
J. Fu, Z. Zhang, G. Li, Chin. Chem. Lett. 30 (2019) 285-291. DOI:10.1016/j.cclet.2018.10.031 |
| [17] |
T.T. Meng, Y.X. Liu, M.T. Liu, et al., Chin. Chem. Lett. 26 (2015) 1179-1182. DOI:10.1016/j.cclet.2015.05.039 |
| [18] |
X. Liu, M. Liu, J. Chen, Z. Li, Q. Yuan, Chin. Chem. Lett. 29 (2018) 1321-1332. DOI:10.1016/j.cclet.2018.03.004 |
| [19] |
C. Chen, D.A. Ridzon, A.J. Broomer, et al., Nucleic Acids Res. 33 (2005) e179. DOI:10.1093/nar/gni178 |
| [20] |
S. Sharbati-Tehrani, B. Kutz-Lohroff, R. Bergbauer, J. Scholven, R. Einspanier, BMC Mol. Biol. 9 (2008) 34-34. DOI:10.1186/1471-2199-9-34 |
| [21] |
S.P. Jonstrup, J. Koch, J. Kjems, RNA 12 (2006) 1747-1752. DOI:10.1261/rna.110706 |
| [22] |
Y. Cheng, X. Zhang, Z. Li, et al., Angew. Chem. Int. Ed. 48 (2009) 3268-3272. DOI:10.1002/anie.200805665 |
| [23] |
H. Liu, L. Li, L. Duan, et al., Anal. Chem. 85 (2013) 7941-7947. DOI:10.1021/ac401715k |
| [24] |
R. Duan, X. Zuo, S. Wang, et al., J. Am. Chem. Soc. 135 (2013) 4604-4607. DOI:10.1021/ja311313b |
| [25] |
X. Zhang, C. Liu, L. Sun, X. Duan, Z. Li, Chem. Sci. 6 (2015) 6213-6218. DOI:10.1039/C5SC02641E |
| [26] |
H. Jia, Z. Li, C. Liu, Y. Cheng, Angew. Chem. Int. Ed. 49 (2010) 5498-5501. DOI:10.1002/anie.201001375 |
| [27] |
K. Wang, K. Zhang, Z. Lv, et al., Biosens. Bioelectron. 57 (2014) 91-95. DOI:10.1016/j.bios.2014.01.058 |
| [28] |
M. Zhou, X. Teng, Y. Li, R. Deng, J. Li, Anal. Chem. 91 (2019) 5295-5302. DOI:10.1021/acs.analchem.9b00124 |
| [29] |
C. Li, Z. Li, H. Jia, J. Yan, Chem. Commun. 47 (2011) 2595-2597. DOI:10.1039/C0CC03957H |
| [30] |
W. Du, M. Lv, J. Li, R. Yu, J. Jiang, Chem. Commun. 52 (2016) 12721-12724. DOI:10.1039/C6CC06160E |
| [31] |
Y. Sun, H. Tian, C. Liu, Y. Sun, Z. Li, Chem. Commun. 53 (2017) 11040-11043. DOI:10.1039/C7CC06140D |
| [32] |
W. Tian, P. Li, W. He, C. Liu, Z. Li, Biosens. Bioelectron. 128 (2019) 17-22. DOI:10.1016/j.bios.2018.12.041 |
| [33] |
E. Koscianska, J. Starega-Roslan, L.J. Sznajder, et al., BMC Mol. Biol. 12 (2011) 14. DOI:10.1186/1471-2199-12-14 |
| [34] |
P.T. Nelson, D.A. Baldwin, L.M. Scearce, et al., Nat. Methods 1 (2004) 155-161. DOI:10.1038/nmeth717 |
| [35] |
T. Notomi, H. Okayama, H. Masubuchi, et al., Nucleic Acids Res. 28 (2000) E63. DOI:10.1093/nar/28.12.e63 |
| [36] |
N. Tomita, Y. Mori, H. Kanda, T. Notomi, Nat. Protoc. 3 (2008) 877-882. DOI:10.1038/nprot.2008.57 |
| [37] |
Y. Mori, T. Notomi, J. Infect. Chemother. 15 (2009) 62-69. DOI:10.1007/s10156-009-0669-9 |
| [38] |
K. Nagamine, T. Hase, T. Notomi, Mol. Cell Probes 16 (2002) 223-229. DOI:10.1006/mcpr.2002.0415 |
| [39] |
R. Wang, R. Zhao, Y. Li, et al., Lab Chip 18 (2018) 3507-3515. DOI:10.1039/C8LC00841H |
| [40] |
K. Hsieh, P.L. Mage, A.T. Csordas, M. Eisenstein, H.T. Soh, Chem. Commun. 50 (2014) 3747-3749. DOI:10.1039/c4cc00540f |
| [41] |
S. Xie, Y. Yuan, Y. Song, et al., Chem. Commun. 50 (2014) 15932-15935. DOI:10.1039/C4CC06449F |
| [42] |
L.J. Xie, X.T. Yang, R.L. Wang, et al., Angew. Chem. Int. Ed. 58 (2019) 5028-5032. DOI:10.1002/anie.201900901 |
| [43] |
L.J. Xie, R.L. Wang, D. Wang, L. Liu, L. Cheng, Chem. Commun. 53 (2017) 10734-10737. DOI:10.1039/C7CC05544G |
| [44] |
L. Cheng, K.G. Abhilash, R. Breslow, Proc. Natl. Acad. Sci. U. S. A. 109 (2012) 12884-12887. DOI:10.1073/pnas.1210846109 |
| [45] |
L. Cheng, A. Mahendran, R.L. Gonzalez Jr., R. Breslow, Proc. Natl. Acad. Sci. U. S. A. 111 (2014) 7920-7924. DOI:10.1073/pnas.1407295111 |
| [46] |
L. Cheng, C. Doubleday, R. Breslow, Proc. Natl. Acad. Sci. U. S. A. 112 (2015) 4218-4220. DOI:10.1073/pnas.1503739112 |
| [47] |
L. Li, L. Yuan, J. Luo, et al., Clin. Exp. Med. 13 (2013) 109-117. DOI:10.1007/s10238-012-0186-5 |
| [48] |
C. Eichelser, D. Flesch-Janys, J. Chang-Claude, K. Pantel, H. Schwarzenbach, Clin. Chem. 59 (2013) 1489-1496. DOI:10.1373/clinchem.2013.205161 |
| [49] |
G. Misso, M.T. Di Martino, G. de Rosa, et al., Mol. Ther. Nucleic Acids 3 (2014) e194. DOI:10.1038/mtna.2014.46 |
| [50] |
D. Lodygin, V. Tarasov, A. Epanchintsev, et al., Cell Cycle 7 (2008) 2591-2600. DOI:10.4161/cc.7.16.6533 |