 2020, Vol. 31
2020, Vol. 31 


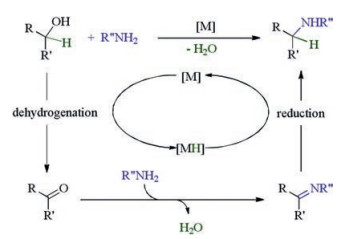
Alcohol amination through the concept of borrowing hydrogen (BH) or hydrogen autotransfer (HA) mediated by homogenous metal catalysis [1], pioneered by Grigg [2] and Watanabe [3] groups, advanced by the groups of Beller [4], Fujita and Yamaguchi [5], Williams [6], Yus [7], Kempe [8], Milstein [9], Deng [10], Xu [11], Shi [12] and others [13], is emerging as an invaluable strategy because it provides a clean and atom economic access [14] to aliphatic amines, a class of important bulky chemicals. This overall redox-neutral process begins with hydrogen transfer from alcohol to metal catalyst [M] to generate related carbonyl compound and metal-hydride complex [MH], condensation of the nascent aldehyde or ketoneRCOR' with primary amine R"NH2 gives imine intermediate which is reduced to alkylated amine by the previously formed metal-hydride [MH] to complete a catalytic circle with water as the sole byproduct (Scheme 1). The almost ideal green methodology has drawn great attention from industry and swiftly found elegant application in pharmaceutic manufacture [15].

|
Download:
|
| Scheme 1. Redox-neutral amination via borrowing hydrogen or hydrogen autotransfer protocol. | |
Simple N-heterocycles can also be made from primary amines through two fold hydrogen autotransfer amination of appropriate diols (Scheme 2a). Butanediol, pentanediol, hexanediol and even heptanediol were all reported for this purpose to achieve corresponding pyrrolidines, piperidines, azepanes and azocanes in various catalytic systems [16]. N-Substituted morpholine and piperazine were accomplished from related diols and primary amines as well [6b, 17]. Notably, the formation of 2-methyl pyrrolidine/piperidine 3 demonstrates that diols bearing one secondary OH group can also participate in this BH process [18]. Based on these precedents, we envisioned it would be remarkable if triols are feasible for this reaction, because precious aliphatic N-heterocycles hydroxylated on the saturated ring would be directly formed. The additional free hydroxyl group could cause huge challenge as it might also involve in and therefore complicate the reaction. Here we would like to report our initial findings at this direction (Scheme 2b).

|
Download:
|
| Scheme 2. Borrowing hydrogen strategy for the synthesis of (a) N-heterocycles from diols and (b) hydroxylated N-heterocycles from triols. | |
3-Pyrrolidinol is a privileged N-heterocycle that finds significant applications in pharmaceutical industry [19]. Its preparation involves multiple-step transformations and/or hazard reagents [20], and an efficient and clean process is highly appreciated. We therefore set out to explore the BH alkylation cyclization of primary amine with 1, 2, 4-butanetriol, which would leading to N-substituted 3-pyrrolidinol derivatives. Realizing that transition metal catalyst plays a key role in hydrogen autotransfer reactions, we started this research with catalyst evaluation. A mixture of aniline 5a with 2 equiv. butanetriol 6a in reflux toluene was used as model reaction to evaluate metal complexes easily accessible in our laboratory in catalytic amount (5 mol%) (Table 1, for experimental details please see Supporting information). Iridium complex [IrCp*Cl2]2, [Ir(cod)Cl2]2 and IrCp*Pro [16e] exhibited no catalytic activity as no consumption of starting aniline occurred (entries 1–3). Although iron cyclone complex FeCyn(CO)3, upon activation by oxidant Me3NO, catalyzed the BH amination of aniline with benzyl alcohol efficiently [21], the same catalyst combination lost its activity completely in current triol amination (entry 4). [RuCp*Cl2]2 and Ru(bby)3Cl2/dppf also failed to promote the desired double N-alkylation (entries 5 and 6). To our delight, catalytic amount of RuHCl(CO)(PPh3)3/XantPhos combination was able to realize the target reaction in the presence of 20 mol% Cs2CO3 and cyclic aminoalcohol 4a was achieved in 58% isolated yield after 48 h reaction and [Ru(p-cymene)Cl2]2 delivered similar outcomes with K2CO3 as the base (entries 7 and 8). Bidentate phosphorines dppf, BINAP and dppBz were much inferior to XantPhos as ligand for [Ru(p-cymene)Cl2]2(entries 9–11), and monophosphine ligands XPhos and DavePhos behaved even worse (entries 12 and 13). These observations underline the importance of ligand effect on this reaction. Using Et3N or t-BuOK as the base instead resulted in decreased yields (entries 14 and 15), whereas Cs2CO3 improve the yield of 4a to 81% under the identical conditions (entry 16). Introduction of 10 mol% AgNO3 as additive didnot improve but unexpectedly ruined the reaction (entry 17). Dioxane as reaction medium worked similarly well as toluene (entry 18). It was found that both 1.5 equiv. and 2.5 equiv. butanetriol provided lower yield than 2.0 equiv. triol could provide (entries 19–20). A slower reaction companied by a lower yield (53%) was observed when the catalyst load was decreased to 2 mol% (entry 21). It was worthy to note that in most cases where low yields were obtained, the starting aniline was not consumed completely. Obviously, the conditions used in entry 16 were the optimal ones and were set as the standard for next studies.
|
|
Table 1 Conditions optimization.a |

{kind=link}
{kind=link}
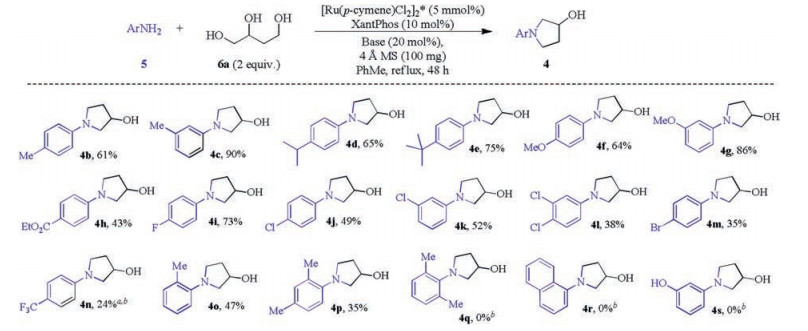
With the optimal and standard conditions in hand, the reaction of a variety of aryl amines with butanetriols 6a were carried out (Scheme 3). N-Aryl-3-pyrrolidinols 4b–4e were produced in mediate to high yields from the two fold N-alkylation reaction of corresponding para or meta alkyl anilines. para and meta methoxyl anilines were smoothly transformed into 4f and 4g in 64% and 86% yield respectively with complete conversion in 48 h. p-CO2Et aniline was also feasible substrate and was successfully converted to 4h, albeit in a slightly decreased yield of 43%. The reaction of 4-F aniline proceeded smoothly to provide pyrrolidinol 4i in 73% yield. N-Aryl-3-pyrrolidinols 4j–4m with Cl or Br substitution were also achieved in moderate yields indicating that these two venerable halidesubstitutions on the aniline are compatible with this transition metal catalyzed process in reflux toluene although relative complicated reaction mixtures were observed. Interestingly, p-CF3 5n aniline was partially transformed into 4n (24% yield) with ca. 70% recovery of starting aniline even after 3 days reaction. The reactions to form 4o and 4p were pretty slower than previous ones and significant amount of starting anilines 5o and 5p remained after 48 h. It can reasonably be attributed to the steric effects and was further confirmed by the entire failure of reactions of the triol with 2, 6-dimethylaniline 5q and 1-naphylamine 5r. m-OH aniline 5s was also inert for this reaction, apparently the phenolic OH deactivated the catalytic system.

|
Download:
|
| Scheme 3. Amination of 1, 2, 4-butanetriol with primary arylamines. Reaction conditions: 5b-q (0.5 mmol), 6a (1.0 mmol), [Ru(p-cymene)Cl2]2 (5 mol%), XantPhos (10 mol%), Cs2CO3 (20 mol%), toluene (2.0 mL), 4 Å MS (100 mg), N2, reflux, sealed tube, 48 h. Isolated yield. a72 h. bStarting anilines was recovered. | |
{kind=link}
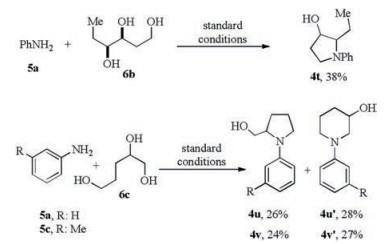
Next, to further test the potential of this catalytic system, 1, 3, 4-hexanetriol 6b was submitted to the standard conditions with aniline 5a as the alkylation acceptor. Pleasingly, 2-ethyl pyrrolidinol 4t was also produced, albeit in only 38% yield (Scheme 4, top). When 1, 2, 5-pentanetriol 6c was used as the alkylating agent, the same reaction with aniline 5a gave rise to a pyrrolidine/piperidine pair 4u/4u' in good combined yield and equal selectivity. Analogous results were obtained for m-Me aniline 5c (Scheme 4, bottom). Related experiment details, characteristics of compounds, copies of NMR spectra can be found in Supporting information for this article.

|
Download:
|
| Scheme 4. Amination cyclization of other two triols. | |
{kind=link}
In summary, a ruthenium catalyzed hydrogen autotransfer amination of triols has been developed for 3-pyrrolidinol synthesis. This catalytic system is proved to be compatible with the additional free hydroxyl group. A variety of substituted anilines are successfully transferred into highly valuable 3-pyrrolidinol derivatives from abound and cheap 1, 2, 4-butanetriol with high efficiency. This extremely environmental benign and low-cost protocol would stimulate further investigations.
AcknowledgmentsWe thank the National Natural Science Foundation of China (No. 21672027), QingLan Project of Jiangsu Province (2016) and Six-Talent-Peaks Program of Jiangsu (2016) for financial support. This work was also supported by High-Level Entrepreneurial Talent Team of Jiangsu Province (No. 2017-37).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.05.027.
| [1] |
(a) T. Irrgang, R. Kempe, Chem. Rev. 119 (2019) 2524-2549; (b) A. Corma, J. Navas, M.J. Sabater, Chem. Rev. 118 (2018) 1410-1459; (c) X. Ma, C. Su, Q. Xu, Top. Curr. Chem. 374 (2016) 1-74; (d) Q. Yang, Q. Wang, Z. Yu, Chem. Soc. Rev. 44 (2015) 2305-2329. |
| [2] |
(a) S. Whitney, R. Grigg, A. Derrick, A. Keep, Org. Lett. 9 (2007) 3299-3302; (b) C. Loefberg, R. Grigg, M.A. Whittaker, A. Keep, A. Derrick, J. Org. Chem. 71 (2006) 8023-8027; (c) R. Grigg, T.R.B. Mitchell, S. Sutthivaiyakit, N. Tongpenyai, Chem. Commun. (1981) 611-612. |
| [3] |
Y. Watanabe, Y. Tsuji, Y. Ohsugi, Tetrahedron Lett. 22 (1981) 2667-2670. DOI:10.1016/S0040-4039(01)92965-X |
| [4] |
(a) S. Elangovan, J. Neumann, J.B. Sortais, et al., Nat. Commun. 7 (2016) 12641; (b) S. Baehn, S. Imm, K. Mevius, et al., Chem. Eur. J. 16 (2010) 3590-3593; (c) S. Imm, S. Baehn, L. Neubert, H. Neumann, M. Beller, Angew. Chem. Int. Ed. 49 (2010) 8126-8129. |
| [5] |
(a) R. Kawahara, K.I. Fujita, R. Yamaguchi, Adv. Synth. Catal. 353 (2011) 1161-1168; (b) R. Kawahara, K.I. Fujita, R. Yamaguchi, J. Am. Chem. Soc. 132 (2010) 15108-15111. |
| [6] |
(a) O. Saidi, A.J. Blacker, M.M. Farah, S.P. Marsden, J.M.J. Williams, Chem. Commun. 46 (2010) 1541-1543; (b) O. Saidi, A.J. Blacker, G.W. Lamb, et al., Org. Process Res. Dev. 14 (2010) 1046-1049. |
| [7] |
(a) R. Martinez, D.J. Ramon, M. Yus, Org. Biomol. Chem. 7 (2009) 2176-2181; (b) R. Martinez, G.J. Brand, D.J. Ramon, M. Yus, Tetrahedron Lett. 46 (2005) 3683-3686. |
| [8] |
(a) S. Michlik, R. Kempe, Chem. Eur. J. 16 (2010) 13193-13198; (b) B. Blank, R. Kempe, J. Am. Chem. Soc. 132 (2010) 924-925. |
| [9] |
C. Gunanathan, D. Milstein, Angew. Chem. Int. Ed. 47 (2008) 8661-8664. DOI:10.1002/anie.200803229 |
| [10] |
(a) S. Liu, R. Chen, G.J. Deng, Chem. Lett. 40 (2011) 489-491; (b) Y. Liu, W. Chen, C. Feng, G. Deng, Chem. Asian J. 6 (2011) 1142-1146. |
| [11] |
S.L. Feng, C.Z. Liu, Q. Li, X.C. Yu, Q. Xu, Chin. Chem. Lett. 22 (2011) 1021-1024. DOI:10.1016/j.cclet.2011.03.014 |
| [12] |
(a) X. Cui, F. Shi, Y. Zhang, Y. Deng, Tetrahedron Lett. 51 (2010) 2048-2051; (b) X. Cui, F. Shi, M.K. Tse, et al., Adv. Synth. Catal. 351 (2009) 2949-2958. |
| [13] |
(a) B. Emayavaramban, P. Chakraborty, E. Manoury, R. Poli, B. Sundararaju, Org. Chem. Front. 6 (2019) 852-857; (b) C.M. Hsiao, Y.F. Chen, C.H. Lin, et al., J. Organomet. Chem. 861 (2018) 10-16; (c) G. Zhang, Z. Yin, S. Zheng, Org. Lett. 18 (2016) 300-303; (d) T. Yan, B.L. Feringa, K. Barta, ACS Catal. 6 (2016) 381-388; (e) Q. Zou, C. Wang, J. Smith, D. Xue, J. Xiao, Chem. Eur. J. 21 (2015) 9656-9661; (f) Y. Zhang, X. Qi, X. Cui, F. Shi, Y. Deng, Tetrahedron Lett. 52 (2011) 1334-1338; (g) J.Y. Zhang, X. Huang, Q.Y. Shen, J.Y. Wang, G.H. Song, Chin. Chem. Lett. 29 (2018) 197-200; (h) T.T. Zhang, J.Y. Jiang, Y.H. Wang, Chin. Chem. Lett. 28 (2017) 307-311. |
| [14] |
C. Gunanathan, D. Milstein, Science 341 (2013) 249. |
| [15] |
(a) J. Leonard, A.J. Blacker, S.P. Marsden, et al., Org. Process Res. Dev. 19 (2015) 1400-1410; (b) M.A. Berliner, S.P.A. Dubant, T. Makowski, et al., Org. Process Res. Dev. 15 (2011) 1052-1062. |
| [16] |
(a) T. Yan, B.L. Feringa, K. Barta, Nat. Commun. 5 (2014) 5602; (b) A.B. Enyong, B. Moasser, J. Org. Chem. 79 (2014) 7553-7563; (c) G. Cami-Kobeci, J.M.J. Williams, Chem. Commun. (2004) 1072-1073; (d) X. Cui, X. Dai, Y. Deng, F. Shi, Chem. Eur. J. 19 (2013) 3665-3675; (e) A. Wetzel, S. Woeckel, M. Schelwies, et al., Org. Lett. 15 (2013) 266-269; (f) M.H.S.A. Hamid, C.L. Allen, G.W. Lamb, et al., J. Am. Chem. Soc. 131 (2009) 1766-1774; (g) I. Yamaguchi, T. Sakano, H. Ishii, K. Osakada, T. Yamamoto, J. Organomet. Chem. 584 (1999) 213-216; (h) D. Seyferth, R.C. Hui, J. Org. Chem. 50 (1985) 1985-1987; (i) L.Y. Xie, S. Peng, L.L. Jiang, et al., Org. Chem. Front. 6 (2019) 167-171. |
| [17] |
(a) A.J.A. Watson, A.C. Maxwell, J.M.J. Williams, J. Org. Chem. 76 (2011) 2328-2331; (b) R.A.T.M. Abbenhuis, J. Boersma, G. van Koten, J. Org. Chem. 63 (1998) 4282-4290. |
| [18] |
(a) K.O. Marichev, J.M. Takacs, ACS Catal. 6 (2016) 2205-2210; (b) K.I. Fujita, T. Fujii, R. Yamaguchi, Org. Lett. 6 (2004) 3525-3528. |
| [19] |
(a) J. Katz, J. Jewell, J. Jung, et al., PT: WO2010017047A1, 2010; (b) G.M. Bright, PT: WO9952907A1, 1999; (c) A. Naylor, D.B. Judd, D.I.C. Scopes, A.G. Hayes, P.J. Birch, J. Med. Chem. 37 (1994) 2138-2144; (d) L. Li, Q. Chen, X. Xiong, et al., Chin. Chem. Lett. 29 (2018) 1893-1896. |
| [20] |
(a) K. Zong, Bull. Korean Chem. Soc. 26 (2005) 717-718; (b) E.J. Trybulski, R.H. Kramss, R.M. Mangano, H.J. Brabander, G. Francisco, Bioorg. Med. Chem. Lett. 2 (1992) 827-832; (c) A. Dicko, M. Montury, M. Baboulene, Tetrahedron Lett. 28 (1987) 6041-6044; (d) M.M. Bowers Nemia, J. Lee, M.M. Joullie, Synth. Commun. 13 (1983) 1117-1123. |
| [21] |
A.J. Rawlings, L.J. Diorazio, M. Wills, Org. Lett. 17 (2015) 1086-1089. DOI:10.1021/ol503587n |