 2020, Vol. 31
2020, Vol. 31 


b College of Chemistry and Pharmaceutical Engineering, Nanyang Normal University, Nanyang 473061, China;
c School of Food and Chemical Engineering, Beijing Technology and Business University, Beijing 100048, China
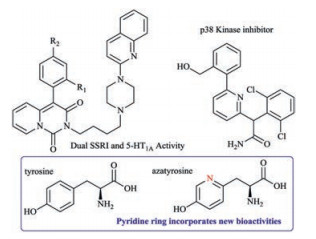
Heterocycles are of great significance to the pharmaceutical industry due to their ability to interact with biological systems through polar interactions [1]. In particular, the unique electronic and conformational properties of pyridine rings renders them one simple yet privileged class of structure motifs in medicinal chemistry (Fig. 1) [2]. The incorporation of pyridine rings often had profound impacts on the biological properties of natural products. For instance, azatyrosine, with replacement of phenyl ring with pyridine, displays potent antibiotic and antitumor properties, when compared to corrsponding tyrosine [3].

|
Download:
|
| Fig. 1. Pyridine motifs in medicinal chemistry. | |
The major progress of synthetic approaches to pyridine-containing molecules relied on the manipulation of pre-functionalized structural motifs [4]. Among those progress, the benzylic functionalization of pyridines is of great importance, as it provides access to useful building blocks such as α-pyridyl carbonyl, germinal dipyridyl moiety [5]. A common procedure for the benzylic functionalization of pyridines involved the deprotonation of picoline precursors with organometallic reagents, followed by the treatment with electrophiles (Scheme 1) [6]. Recently, alternative approaches have also been reported. For instance, Londregan and co-workers reported the PyBroP-mediated the addition of silyl ketene acetals to azine-N-oxides [7]. Nevertheless, these protocols showed broad synthetic appeal and were useful in certain contexts, but the reaction reagent nature still limited their utilities to a certain degree [6, 8].

|
Download:
|
| Scheme 1. Pyridine benzylic functionalization. | |
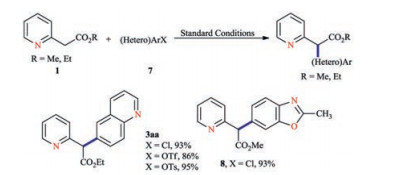
Given our group's continuous efforts in heterocyclic chemistry and transition metal catalysis [[9]], our attention was therefore drawn to employ the Pd-catalyzed α-arylation reaction to achieve the pyridyl-benzylic functionalization. Since the seminal work from Buchwald, Hartwig and Miura groups, the Pd-catalyzed α-arylation of carbonyl moiety has emerged as an effective C(sp2)-C(sp3) bond formation strategy [10]. A wide array of protocols has been found to be effective to connect highly functionalized substrates. Moreover, these progresses have transformed the way in which synthetic chemists designed and implemented synthesis on both bench-top and industrial scales [11]. Notwithstanding such progress, the substrate scope of both coupling partners remains challenging with respect to the pyridine functionality. In principle, any Pd-catalyzed α-arylation reactions that is tolerable to both the Lewis basic functionality as well as the inherent electronic deficient property of pyridine rings, would be highly appreciated, and offer alternative efficient solution to pyridine benzylic functionalization [9a, 12]. Another critical task is to identify the suitable reaction conditions to avoid the inhibition caused by the enolization of 2-(pyridin-2-yl)acetate and resulting coordination with palladium center as the ligand. Despite these challenges, we would like to report a highly efficient pyridyl-benzylic functionalization protocol via the palladium-catalyzed mono-α-arylation reaction of α-(2-azaheteroaryl) acetates with heteroaryl halides. Due to the immense usefulness of pyridine motifs, this protocol in principle should be of great interests to the community.
We initiated the optimization study with commercially available ethyl 2-(pyridin-2-yl)acetate (1a) and 6-bromoquinoline (2a) (Table 1). Among the phosphine ligands examined, the highest yield of ethyl 2-(pyridin-2-yl)-2-(quinolin-6-yl)acetate (3aa) was obtained in the presence of X-Phos (L6) (Fig. 2) [13]. Next, a series of palladium sources were examined, all of which led to slightly lower yields, when compared to palladium acetate. Attention was then switched to determine the optimal solvent for the reaction. The control experiments revealed that dioxane was the ideal choice (entries 8–12). Keeping all other variables constant, the coupling reactions with either Na2CO3 or K3PO4 were then performed, and both failed to offer better results (entry 6 vs. entries 13 and 14).
|
|
Table 1 Optimization of the reaction conditions.a |

{kind=link}
{kind=link}

|
Download:
|
| Fig. 2. Structures of ligands (L1-L6). | |
{kind=link}
With the optimal conditions in hands, the transformation was demonstrated on a range of heteroaryl halides. As shown in Scheme 2, investigations of the heteroaryl bromide scope showed that quinoline-based heteroaryl bromides were viable substrates to give desired products in excellent yields (3aa-3ae, 75%–96% yields). Moreover, pyridine-containing substrates proved to be well-tolerant to give the desired adducts 3af-3am in 83%–97% yields. It is worth mentioning that these substrate classes are discovered to be non-sensitive to the electronic effect of substitution groups on the pyridine rings. Besides, thiophene derived substrates, which are also widely present in medicinally relevant compounds, underwent the reaction smoothly to deliver coupling products (3an, 3bd, 3be, 63%–83% yields). As expected, when simple bromobenzene was employed, the desired arylation product 3ao was obtained in 95% yield.

|
Download:
|
| Scheme 2. Substrate scope. Reaction conditions: 1a (0.3 mmol), 2 (0.4 mmol), Pd(OAc)2 (0.015 mmol), X-Phos (0.015 mmol) and Cs2CO3 (0.9 mmol) in dioxane (1 mL), 100 ℃, N2, 10 h. Isolated yields. | |
{kind=link}
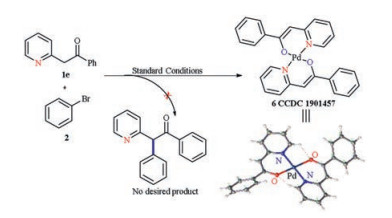
Next, we examined the generality of our coupling protocol with respect to the heteroaryl substituted acetates (Scheme 3). The methyl and t-Bu 2-(pyridin-2-yl)acetate are viable substrates, providing quick access to desired products (3ba-3cg) in good yields (63%–97% yields). We were also glad to discovery that the protocol can be extended to quinolidyl acetates, which performed well under the standard conditions to give 3 dh in 88% yield. More importantly, the mono-arylation product could be utilized to further react with bromocyclopropane to construct the all carbon quaternary center on 5, which is presumably involve β-elimination before coupling [14, 15]. Unfortunately, when we employed 1e, the palladiumcomplex 6 was obtained instead of the corresponding coupling products (Scheme 4). Single crystals suitable for X-ray diffraction analysis were grown from a mixture of dichloromethane and petroleum ether. The central palladium atom displayed a square-planar arrangement with the nitrogen atoms of the pyridine ring and oxygen of the enol moiety situated mutually in the trans position.

|
Download:
|
| Scheme 3. Substrate scope. Reaction conditions: 1 (0.3 mmol), 2 (0.4 mmol), Pd(OAc)2 (0.015 mmol), X-Phos (0.015 mmol) and Cs2CO3 (0.9 mmol) in dioxane (1 mL), 100 ℃, N2, 10 h. Isolated yields. | |
{kind=link}

|
Download:
|
| Scheme 4. Ketone substrate and resulting palladium complex. Crystal data: C26H20N2O2Pd, M = 498.84, triclinic, P1, a = 9.8373(3), b = 12.0510(4), c = 17.9512(6) Å, α = 105.345(3), β = 95.871(3), γ = 93.103(3), V = 2034.16(11) Å3, Z = 4, Mo Kα radiation (λ = 0.71073). For details, see Supporting information. | |
{kind=link}
Alteration of the halide fragments was evaluated next, using heteroaryl chlorides, triflates and tosylates as the coupling partners. Under optimized conditions, the arylated products 3aa were still obtained in good chemical yields, respectively. Additionally, our protocol was effective to incorporate benzothiazole units into arylation products using 5-chloro-2-methylbenzothiazole (Scheme 5). Noteworthy, all the above-mentioned reactions showed no evidence of over-arylation by-products, which is a major side pathway for the arylation reactions of α-carbonyl compounds [16].

|
Download:
|
| Scheme 5. Substrate scope. | |
{kind=link}
This arylation protocol is amenable to gram-scale preparation of 3aa. We reacted 6 mmol of 1a with a slight excess amount (1.3 equiv., 7.8 mmol) of 2a. Under the same reaction conditions, 3aa was produced in 91% yield (Scheme 6). Next, the hydrolysis and subsequent decarboxylation went without issue to provide the diheteroaryl ketone product 10 in good efficiency. This one-pot, two-step protocol was further demonstrated by the quick synthesis of 11 and 12, which often displayed photochemical reactivities [17].

|
Download:
|
| Scheme 6. Synthetic applications. | |
{kind=link}
In conclusion, we have developed an efficient Pd-catalyzed mono-α-arylation reaction of α-(2-azaheteroaryl) ester with heteroaryl halides. The broad heterocyclic substrate scope enabled the rapid synthesis of a wide array of functionalized α, α'-di-heteroaryl acetate products with consistently good yields. Remarkably, our protocol proved to be not only efficient in scale-up reactions, but further functionalization was also readily possible, setting the base for rapidly accessing diheteroaryl ketonederivatives. In comparison to classical approaches for pyridine benzylic functionalization, we anticipate that our catalytic protocol will provide an efficient alternative for divergent parallel synthesis efforts.
AcknowledgmentsTan is grateful to the support by the National Natural Science Foundation of China (No. 21702013), Beijing Natural Science Foundation (No. 2184115) and the Fundamental Research Funds from the Central Universities (Nos. XK1802-6, buctrc201721) in Beijing University of Chemical Technology.
Appendix A. Supplementary dataSupplementary material related to this article can be found, inthe online version, at doi:https://doi.org/10.1016/j.cclet.2019.06.028.
| [1] |
(a) R.E. Dolle, B.L. Bourdonnec, K. Worm, et al., Comb. Chem. 12 (2010) 765-806; (b) A. Facchetti, Chem. Mater. 23 (2011) 733-758; (c) L.M. Blair, J. Sperry, J. Nat. Prod. 76 (2013) 794-812; (d) E. Vitaku, D.T. Smith, J.T. Njardarson, J. Med. Chem. 57 (2014) 10257-10274; (e) U.H.F. Bunz, Acc. Chem. Res. 48 (2015) 1676-1686; (f) R.K. Alan, A.R. Christopher, F.V.S. Eric, et al., Comprehensive Heterocyclic Chemistry Ⅲ, Elsevier, Oxford, 2008; (g) K.C. Majumdar, S.K. Chattopadhyay, Heterocycles in Natural Product Synthesis, Wiley-VCH, Weinheim, 2011. |
| [2] |
(a) J. Alvarez-Builla, J.J. Vaquero, J. Barluenga, Modern Heterocyclic Chemistry, Wiley-VCH, Weinheim, 2011; (b) M. Baumann, I.R. Baxendale, Beilstein J. Org. Chem. (9) (2013) 2265-2319; (c) M.D. Hill, Chem. Eur. J. 16 (2010) 12052-12062; (d) J.S. Carey, D. Laffan, C. Thomson, et al., Org. Biomol. Chem. 4 (2006) 2337-2347; (e) M. Schlosser, F. Mongin, Chem. Soc. Rev. 36 (2007) 1161-1172; (f) L. Ackermann, H.K. Potukuchi, A.R. Kapdi, et al., Chem. Eur. J. 16 (2010) 3300-3303; (g) Y.S. Kumar, F.-R.N. Khan, Chin. Chem. Lett. 28 (2017) 1607-1612; (h) A.P. Krinochkin, D.S. Kopchuk, N.V. Chepchugov, et al., Chin. Chem. Lett. 28 (2017) 1099-1103. |
| [3] |
R.A. Aycock, D.B. Vogt, N.T. Jui, Chem. Sci. 8 (2017) 7998-8003. DOI:10.1039/C7SC03612D |
| [4] |
(a) A. Kotschy, G. Timári, Heterocycles From Transition Metal Catalysis, Springer, Dordrecht, 2005; (b) S. Schröter, C. Stock, T. Bach, Tetrahedron 61 (2005) 2245-2267; (c) M. Beller, C. Bolm, Transition Metals for Organic Synthesis: Building Blocks and Fine Chemicals, 2nd ed., Wiley-VCH, Weinheim, 2004. |
| [5] |
(a) H.-Y. Lin, B.B. Snider, J. Org. Chem. 77 (2012) 4832-4836; (b) J.A. Lowe, D.L. Hageman, S.E. Drozda, et al., J. Med. Chem. 37 (1994) 3789-3811; (c) J.J. Mousseau, A. Larivée, A.B. Charette, Org. Lett. 10 (2008) 1641-1643; (d) S. Duez, A.K. Steib, S.M. Manolikakes, et al., Angew. Chem. Int. Ed. 50 (2011) 7686-7690; (e) K.P. Bogeso, A.V. Christensen, J. Hyttel, et al., J. Med. Chem. 28 (1985) 1817-1828. |
| [6] |
(a) R.B. Woodward, E.C. Kornfeld, Org. Synth. 29 (1949) 44; (b) W.G. Kofron, L.M. Baclawski, Org. Synth. 52 (1972) 75; (c) R. Zhu, G. Cheng, C. Jia, et al., J. Org. Chem. 81 (2016) 7539-7544. |
| [7] |
(a) A.T. Londregan, S. Jennings, L. Wei, Org. Lett. 12 (2010) 5254-5257; (b) A.T. Londregan, S. Jennings, L. Wei, Org. Lett. 13 (2011) 1840-1843. |
| [8] |
(a) P.S. Fier, J. Am. Chem. Soc. 139 (2017) 9499-9502; (b) D.D. Zhai, X.Y. Zhang, Y.F. Liu, et al., Angew. Chem. Int. Ed. 57 (2018) 1650-1653 and references therein. |
| [9] |
(a) J.J. Tan, Y. Chen, H. Li, et al., J. Org. Chem. 79 (2014) 8871-8876; (b) H.Q. Wang, W.T. Xu, Z.Q. Wang, et al., J. Org. Chem. 80 (2015) 2431-2435; (c) L. Liu, C. Tan, R. Fan, et al., Org. Biomol. Chem. 17 (2019) 252-256. |
| [10] |
(a) S. Lee, N.A. Beare, J.F. Hartwig, J. Am. Chem. Soc. 123 (2001) 8410-8411; (b) M. Jorgensen, S. Lee, X. Liu, et al., J. Am. Chem. Soc. 124 (2002) 12557-12565; (c) T. Hama, X. Liu, D.A. Culkin, et al., J. Am. Chem. Soc.125 (2003) 11176-11177; (d) X. Liu, J.F. Hartwig, J. Am. Chem. Soc. 126 (2004) 5182-5191; (e) T. Hama, J.F. Hartwig, Org. Lett. 10 (2008) 1545-1548; (f) M.R. Biscoe, S.L. Buchwald, Org. Lett. 11 (2009) 1773-1775; (g) T. Hama, D.A. Culkin, J.F. Hartwig, J. Am. Chem. Soc.128 (2006) 4976-4985; (h) B. Zheng, T. Jia, P.J. Walsh, Org. Lett. 15 (2013) 4190-4193; (i) R. Martin, S.L. Buchwald, Angew. Chem. Int. Ed. 46 (2007) 7236-7239; (j) G.D. Vo, J.F. Hartwig, Angew. Chem. Int. Ed. 47 (2008) 2127-2130; (k) R. Martin, S.L. Buchwald, Org. Lett. 10 (2008) 4561-4564; (l) D.A. Culkin, J.F. Hartwig, Acc. Chem. Res. 36 (2003) 234-245; (m) F. Bellina, R. Rossi, Chem. Rev. 110 (2010) 3850; (n) C.C.C. Johansson, T.J. Colacot, Angew. Chem. Int. Ed. 49 (2010) 676-707; (o) Z. Liu, M. Li, B. Wang, et al., Org. Chem. Front. 5 (2018) 1870-1876; (p) G. Gao, Y. Fu, M. Li, et al., Adv. Synth. Catal. 359 (2017) 2890-2894; (q) K. Ablajan, G.B. Panetti, X. Yang, et al., Adv. Synth. Catal. 359 (2017) 1927-1932; (r) G. Saini, P. Kumar, G.S. Kumar, et al., Org. Lett. 20 (2018) 441-444; (s) I. Astarloa, R. SanMartin, M.T. Herrero, et al., Adv. Synth. Catal. 360 (2018) 1711-1718; (t) D.J. Leonard, J.W. Ward, J. Clayden, Nature 562 (2018) 105-109. |
| [11] |
(a) Á. Molnár, Palladium-Catalyzed Coupling Reactions-Practical Aspects and Future Developments, Wiley-WCH, Weinheim, 2013; (b) M.L. Crawley, B.M. Trost, Applications of Transition Metal Catalysis in Drug Discovery and Development, Wiley, New York, 2012; (c) C. Torborg, M. Beller, Adv. Synth. Catal. 351 (2009) 3027-3043. |
| [12] |
(a) M.A. Oberli, S.L. Buchwald, Org. Lett. 14 (2012) 4606-4609; (b) P.E. Maligres, J. Li, S.W. Krska, et al., Angew. Chem. Int. Ed. 51 (2012) 9071-9074; (c) J.C. Tellis, D.N. Primer, G.A. Molander, Science 345 (2014) 433-436. |
| [13] |
(a) X. Huang, K.W. Anderson, D. Zim, J. Am. Chem. Soc. 125 (2003) 6653-6655; (b) N.C. Bruno, M.T. Tudge, S.L. Buchwald, Chem. Sci. 4 (2013) 916-920; (c) P. Novak, R. Martin, Curr. Org. Chem. 15 (2011) 3233-3262. |
| [14] |
(a) C.J. Douglas, L.E. Overman, Proc. Natl. Acad. Sci. U. S. A. 101 (2004) 5363-5367; (b) J. Christoffers, A. Baro, Adv. Synth. Catal. 347 (2005) 1473-1482; (c) K.W. Quasdorf, L.E. Overman, Nature 516 (2014) 181-191. |
| [15] |
(a) J. Gu, X. Wang, W. Xue, Org. Chem. Front. 2 (2015) 1411-1421; (b) X. Wang, S.Z. Stankovich, R.A. Widenhoefer, Organometallics 21 (2002) 901-905. |
| [16] |
(a) W.A. Moradi, S.L. Buchwald, J. Am. Chem. Soc. 123 (2001) 7996-8002; (b) T. Hama, J.F. Hartwig, Org. Lett. 10 (2008) 1549-1552; (c) F. Churruca, R. SanMartin, R. Tellitu, Tetrahedron Lett. 44 (2003) 5925-5929; (d) K.B. Urkalan, M.S. Sigman, Angew. Chem. Int. Ed. 48 (2009) 3146-3149; (e) F. Churruca, R. SanMartin, M. Carril, Tetrahedron 60 (2004) 2393-2408. |
| [17] |
G. Favaro, F. Ortica, A. Romani, J. Photochem. Photobiol. C 16 (2013) 22-45. DOI:10.1016/j.jphotochemrev.2013.03.001 |