 2020, Vol. 31
2020, Vol. 31 

b College of Chemistry and Pharmaceutical Sciences, Qingdao Agricultural University, Qingdao 266109, China;
c School of Chemistry and Chemical Engineering, Hunan University of Science and Technology, Xiangtan 411201, China
Organosulfur compounds exist extensively in nature [1], and also have wide application in a range of fields such as drug development [2], organic material [3], ligand design [4] or polymer science [5]. As a consequence, the development of efficient synthetic methods for the C-S bond formation has gained significant interest among medicinal and synthetic organic chemists over the past few decades [6]. Traditionally, transition-metal-catalyzed cross-couplings aining reagents is well known [7]. In spite of some great advantages, some of these processes could sbetween C-X/C-H and different sulfenylating sources such as thiols, sulfonyl chlorides, disulfides, sulfonyl hydrazides, and other S-contuffer from certain drawbacks, including pre-functionalization of reactants, harsh reaction conditions, and toxic metal catalysts.
With the increased awareness of environmental protection and development of green chemistry, therefore metal-free sulfenylation via direct functionalization of C-H bonds have attracted much attention in recent years and there are more and more excellent and significant works have been presented. Moreover, several reviews on incorporation of mercapto into molecules with sundry sulfur reagents from various views [8]. This review will focus on the recent five-year advances in C-S bond formation via direct sulfenylation of C(sp3)H bonds under metal-free conditions and elaborate their mechanisms from a new perspective.
2. Non-enantioselective sulfenylation of C(sp3)H 2.1. 1, 3-Dicarbonyl compoundsAs the crucial starting material in organic synthesis, the embellishments of 1, 3-dicarbonyl compounds are always the focus in chemical research. Among them, the sulfenylation of the methylene site of 1, 3-dicarbonyl compounds has aroused great interest from the scientific community, since sulfur-containing motifs are significant in both synthetic chemistry and medicinal chemistry [9]. However, sulfenylation of β-diketones is still challenging as β-diketones are apt to undergo deacylation after sulfenylation in the reaction medium.
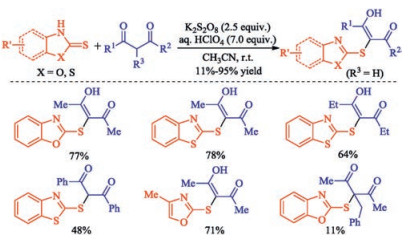
In 2015, Prabhu group reported the sulfenylation of β-diketones without deacylation via a cross dehydrogenative coupling (CDC) strategy [10]. α-Sulfenylated β-diketones could be obtained utilizing heteroarylthiones as a thiol equivalent under metal-free conditions at ambient temperature. It is noteworthy that the sulfenylated β-diketones exist primarily in the form of enol, but predominantly existed in their keto form when using α-substituted β-diketone as substrate or incorporation more steric hindrance group R (Scheme 1).

|
Download:
|
| Scheme 1. Sulfenylation of β-diketones with heteroarylthiones. | |
{kind=link}
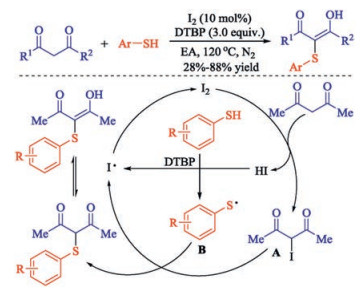
A novel method on iodine-catalyzed sulfenylation of 1, 3-diketones with thiophenols to form β-dicarbonyl thioethers was developed by Hu and co-workers [11]. Various symmetrical sulfenylated 1, 3-dicarbonyl compounds were obtained under metal free mild conditions in moderate to excellent yields with good functional group tolerance. The C-S bond was supposed to be formed via a radical substitution pathway instead of the usual nucleophilic substitution one. The diketone is firstly iodinated by iodine catalyst to form the key intermediate A and the thiyl radical B is formed from oxidation of thiophenol by DTBP. Then radical B would attack the unstable C-I bond of A. Through a radical substitution pathway, the β-dicarbonyl thioether product was formed. Finally, the HI released from iodination is oxidized by DTBP to regenerate the iodine catalyst (Scheme 2).

|
Download:
|
| Scheme 2. Sulfenylation of β-diketones with thiophenols. | |
{kind=link}
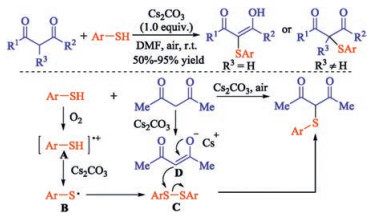
This kind of sulfenylation of β-diketones with thiophenols also can underwent well using potassium iodide and oxygen as the equivalent of iodine to catalyze. For instance, Wang group demonstrated a practical methodology to obtain α-thio-β-dicarbonyl compounds using potassium iodide (KI) as catalyst [12]. The presented environmentally benign access provides both symmetrical and unsymmetrical α-thio-β-dicarbonyl compounds in good yields under an aerobic atmosphere. Subsequently, Chen and co-workers found a convenient Cs2CO3-promoted α-sulfenylation of β-diketones under halogen-free conditions [13]. This transformation provides a straightforward route to α-sulfenylated β-dicarbonyl compounds using air as the oxidant under mild conditions with wide functional group compatibility. A plausible reaction mechanism is outlined in Scheme 3. Initially, thiyl radical is generated from the autoxidation of thiol in the presence of Cs2CO3 and oxygen, and then undergoes homocoupling to produce disulfide. Meanwhile, β-diketones reacts with Cs2CO3 to form intermediate D. Finally, the nucleophilic attack of the in situ generated enolate D on disulfide C affords α-sulfenylated carbonyl compound (Scheme 3).

|
Download:
|
| Scheme 3. Cs2CO3-Promoted sulfenylation of β-diketones. | |
{kind=link}
In 2015, Lee and co-workers developed an interesting transition metal free K2S2O8/I2 promoted sulfenylation of β-diketones with disulfides at room temperature under solvent free conditions [14]. Both diaryl and dialkyl disulfides are coupled well with a variety of β-diketones providing α-thio-β-diketones in good to excellent yields. Authors proposed a plausible mechanism for this transformation as shown in Scheme 4. In presence of I2, disulfides can attack on dicarbonyl compounds to provide the desired product along with the formation of HI and R3SI (A) as shown in Path A. Alternatively, the enol form of dicarbonyl compounds can also undergo a similar kind of reaction to generate product along with HI and R3SI (Path B). Moreover, the enol form of dicarbonyl compounds also can react with very reactive species A to provide the desired product according to Path C. Finally, HI can be reoxidized into molecular iodine in presence of oxidant (Scheme 4). Barman group found that additional alkali could accelerate the sulfenylation of β-diketones with disulfides. They firstly reported a TBATB and Et3N mediated protocol for sulfenylation of active methylene compounds with diaryl disulfide [15]. This new process is not air or moisture sensitive and afforded the desired products in excellent yields. Additionally, the same group developed the sulfenylation of active methylene compounds promoted by iodine, Et3N and DMSO under mild conditions [16]. Selective mono- and bis-sulfenylation of active methylene groups with a variety of disulfides at an ambient temperature was realized by vary of the amount of DMSO and base. The present work has more substrate scope and is greener in terms of solvent selection and the use of less hazardous DMSO as an oxidant.

|
Download:
|
| Scheme 4. Sulfenylation of β-diketones with disulfides. | |
{kind=link}
The plausible mechanism was assumed in Scheme 5 that shows two catalytic cycles. In the first cycle, the active methylene group easily losses active H in the presence of Et3N and binds to the sulfenyl iodide in situ generated from disulfide to form monosulfenylated product and HI. The catalyst I2 is regenerated by oxidation of HI. However, if the amounts of the intermediate RSI, Et3N and DMSO are sufficiently large, the reaction selectively affords disulfenylated product via a similar pathway at high yield. Quenching of acid by base does not occur because the addition of base promotes the reaction by abstracting the active proton, whereas its absence leads to a very slow process with lower levels of conversion (Scheme 5).

|
Download:
|
| Scheme 5. Et3N-Promoted sulfenylation of β-diketones with disulfides. | |
{kind=link}
2.2. α-H of carbonyl compounds
The chemistry of β-ketosulfides has always been the subject of intense research efforts, because they are very useful building blocks or intermediates for further transformations in organic synthesis [17]. Thus, many researchers have devoted tremendous efforts to develop highly efficient protocols towards C-S bond formation methods. But among these methods, the direct sulfenylation of ketones remains a puzzle as the ketones are not reactive enough compared to β-diketones for α-sulfenylation.
Recently, Yu and co-workers successfully developed a direct oxidative disulfenylation/cyclization of 2-hydroxyacetophenones with thiophenols mediated by TBAI/K2S2O8 [18]. The reaction provides a novel approach for the synthesis of functionalized 2, 2-dithio-benzofuran-3(2H)-ones in good yield. In a single step, two C-S bonds and one C-O bond are simultaneously built on the same sp3 carbon atom under transition metal-free conditions. Meanwhile, a plausible reaction mechanism through two single electron-transfer processes followed by intramolecular cyclization has been proposed. Initially, three intermediates thiyl radical A, disulfide B and RSI C can be generated from thiophenols in the presence of TBAI/K2S2O8. Next, the sulfenylated enol intermediate G was formed from the enol intermediate D of 2-hydroxyacetophenones via the α-sulfenylated carbon radical intermediate E or sulfenylated intermediate F. Then, the second single electron-transfer process is completed by the free radical addition of G with A and followed by a deprotonation process, leading to the disulfenylated enol intermediate I formation. The final intramolecular cyclization of the disulfenylated enol intermediate K results in the desired 2, 2-dithio-benzofuran-3(2H)-one products (Scheme 6).

|
Download:
|
| Scheme 6. Disulfenylation/cyclization of 2-hydroxyacetophenones. | |
{kind=link}
In 2016, Prabhu group achieved a metal-free regioselective sulfenylation of the α-CH3 group of ketones in the presence of the α-CH2 or α-CH group using the cross dehydrogenative strategy [19]. This efficient sulfenylation of ketones with thiones or thiols utilizes dimethyl sulfoxide as an oxidant in the presence of iodine, and aldehydes also exhibit good selectivity forming the corresponding α-sulfenylated products. It is noteworthy that this is the first report of the regioselective sulfenylation of methyl ketones in the presence of α-CH2 or α-CH groups and aldehydes.
A tentative mechanism is proposed in Scheme 7. Thiones tautomerize to thiol in acidic condition. Iodine reacts with thiols furnishing disulfides A. Disulfides further reacts with iodine to form an intermediate ArSI. Nucleophilic displacement of the iodo group of intermediate B from the enolate of ketone or aldehyde furnishes α-sulfenylated product. DMSO reacts with HI to regenerate catalyst I2 (Scheme 7).

|
Download:
|
| Scheme 7. Sulfenylation of the α-CH3 group of ketones. | |
{kind=link}
On the basis of this work, the same group also described a rare regioselective sulfenylation of α′-CH3 or α′-CH2 bonds adjacent to α, β-unsaturated ketones without undergoing conjugate addition [20] and the synthesis of sulfenylated pyrazoles using the same catalytic system (Scheme 8) [21]. The sulfenylation using strong conjugate acceptors such as α, β-unsaturated ketones as nucleophiles exhibits a high regioselectivity without undergoing conjugate addition, which is difficult to achieve under the cross dehydrogenative coupling method. This is a rare phenomenon and is unprecedented.

|
Download:
|
| Scheme 8. Sulfenylation of α, β-unsaturated ketones and pyrazolones. | |
{kind=link}
However, Barman and co-workers found if using base and iodine to promote the sulfenylation, the reaction regioselectivity is controlled to give substituted β-ketosulfides solely at the more hindered carbon, which is a thermodynamically more stable enolate generated product [22]. This synthetic procedure has never been reported and thus significantly attractive. The proposed mechanism is similar to the aforementioned. The progress of the reaction also involves the intermediate RSI followed by attack by the highly substituted carbon center of unsymmetrical ketone to give product, just enolate at the more hindered carbon is more stable under the action of potassium t-butoxide affording the corresponding products. Finally, the iodide ions are reoxidized to iodine molecule in presence of DMSO and solvent to complete the catalytic cycle (Scheme 9).

|
Download:
|
| Scheme 9. Sulfenylation of the α-CH3 group of ketones under KtOBu. | |
{kind=link}
Significant efforts also have been made to synthesize sulfenylated pyrazoles due to their extensive application in various disciplines like materials chemistry, organic synthesis, bio- and medicinal chemistry possessing a broad spectrum of biological activities [23]. In 2017, Kamani group developed a novel strategy for N-chlorosuccinimide mediated direct sulfenylation of pyrazolones using aryl thiols as sulfur source at room temperature [24]. The presented protocol afforded numerous sulfenylated pyrazole derivatives in good to excellent yields without further purification. A plausible reaction mechanism for the transformation has been proposed in Scheme 10. First, the reaction of NCS with thiophenol forms the intermediate A. The subsequent nucleophilic attack of C-4 of pyrazole ring on A generates the intermediate B followed by dehydrochlorination. Finally, rearomatization of B leads to the formation of the desired product by the formation of favored tautomer (Scheme 10).

|
Download:
|
| Scheme 10. NCS-Mediated sulfenylation of pyrazolones. | |
{kind=link}
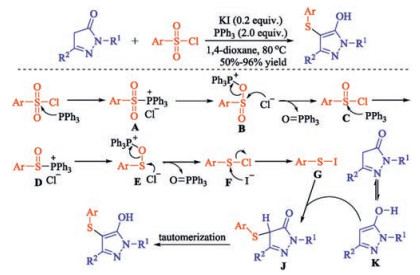
The direct sulfenylation of pyrazolones and oxindoles using N-(thio)succinimides or sulfonyl chlorides as sulfenylating reagents was also described by Zhao et al. [25]. For instance, Zhao and co-workers developed an efficient method to synthesize pyrazolone thioethers by employing aryl sulfonyl chlorides as sulfenylating reagents [25b]. Potassium iodide was found to facilitate the transformation for the first time by generating more reactive sulfenyl iodide in situ from sulfenyl chloride in the presence of triphenylphosphine. Initially, aryl sulfonyl chloride is reduced by PPh3 via intermediates A-E to sulfenyl chloride F, which is attacked by iodide anion to give sulfenyl iodide G. Electrophilic thiolation of pyrazolone tautomer K by sulfenylating reagent G gives the corresponding pyrazolone thioethers (Scheme 11). The protocol also can provide 2-aryl, and 3-aryl benzofuran thioethers under the same conditions.

|
Download:
|
| Scheme11. Sulfenylation of pyrazolones with sulfonyl chlorides. | |
{kind=link}
The direct sulfenylation of the α-CH of ketones also proceeded smoothly under both transition metal and halogen free conditions. A convenient NaOH-promoted direct sulfenylation of pyrazolones with aryl thiols under mild conditions was demonstrated by Wang and co-workers [26]. In this transformation, a range of valuable sulfenylated pyrazoles can be easily achieved in moderate to excellent yields, with high atom efficiency and good functional group tolerance. The study of mechanism shows that first, the hydrogen abstraction of pyrazolone by NaOH would produce carbanion intermediate A, which underwent the rapid tautomerization leading to the enolate anion intermediate B. Subsequently, the nucleophilic substitution of B with disulfide that generated from thiol gave the corresponding sulfenylated pyrazolone C. Finally, the rapid tautomerization of C would lead the generation of the desired product (Scheme 12).

|
Download:
|
| Scheme 12. NaOH-Promoted sulfenylation of pyrazolones. | |
{kind=link}
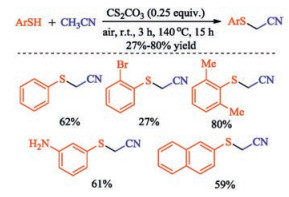
In 2017, Chen group achieved halogen-free Cs2CO3-promoted cross dehydrogenative coupling (CDC) of thiophenols with acetonitrile. The proposed mechanism resembled Wang's work that the carbanion produced via hydrogen abstraction of acetonitrile by Cs2CO3 nucleophilic attack on disulfide that generated from thiol. This transformation provides a straightforward route to the synthesis of a wide range of sulfenylated acetonitriles in up to 80% yield (Scheme 13) [27]. In addition, a wide range of other α-sulfenylated ketones including cycloketones, aldehydes and lactones were also synthesized via halogen-free sulfenylation under eco-friendly conditions [28].

|
Download:
|
| Scheme 13. Cs2CO3-promoted sulfenylation of acetonitriles. | |
{kind=link}
2.3. Inert CH of alkanes
Although there are many excellent works on the sulfenylation of active CH bonds of ketones to be achieved, researches about the sulfenylation of inert CH bonds of alkanes are rare. In 2016, Wu group performed a concise pathway to the sulfenylation of cycloalkanes with alkyl arylsulfinates [29]. A wide range of thioethers were produced in moderate to high yields with iodine as the catalyst through inert C(sp3) H sulfenylation. Notably, this is the first report on using alkyl arylsulfinates as the sulfenylating agent in cross-coupling transformation.
Authors proposed a reaction mechanism as shown in Scheme 14. The reaction started with a thermal homolytic cleavage of a peroxide to generate the t-butoxy radical, which reacts with cyclohexane to provide radical A. Meanwhile, alkyl arylsulfinates is reduced by diethyl phosphonate and I2 to generate the aryl thiol radical B, which is considered as the resting state of phenyl disulfide. Finally, A reacts with B to afford the final product thioethers (Scheme 14).

|
Download:
|
| Scheme 14. Sulfenylation of cycloalkanes with alkyl arylsulfinates. | |
{kind=link}
In 2018, Wang and co-workers disclosed the first protocol for the sulfenylation of the unactivated methyl C(sp3)-H bond in 2-picolines [30] (Scheme 15). The protocol comprises a one-pot, two-step reaction through a TFAA-mediated [3, 3]-sigmatropic rearrangement of pyridine N-oxides and TBAB-catalyzed direct conversion of trifluoroacetates into thioethers under metal- and base-free conditions. The present method is mild, scalable, highly regioselective, and step- and atom-economical. The TBAB catalyst is not required for those highly electron-rich 2-picoline N-oxides. On the other hand, the method doesnot work well for electron-poor N-oxides or alkylthiols.

|
Download:
|
| Scheme 15. Sulfenylation of 2-picolines with thiols. | |
{kind=link}
Recently, Chen and co-workers described the highly efficient synthesis of 9-thiolated fluorenes through a direct base-promoted sulfenylation of fluorenes with thiols [31]. This reaction occurs at ambient conditions and shows good tolerance of functional groups including arylthiols and alkylthiols. A wide range of products are obtained in good yields. A putative mechanism is outlined in Scheme 16 for this transformation. Initially, phenthiol is oxidized by TBHP to produce the intermediate A. Meanwhile, the fluorine was abstracted a proton by base to furnish the anion B, which then undergoes a nucleophilic attack on A to the target product and releases the anion C. Finally, B can be transformed to the intermediate A in the presence of oxidant (Scheme 16).

|
Download:
|
| Scheme 16. Sulfenylation of fluorines with thiols. | |
{kind=link}
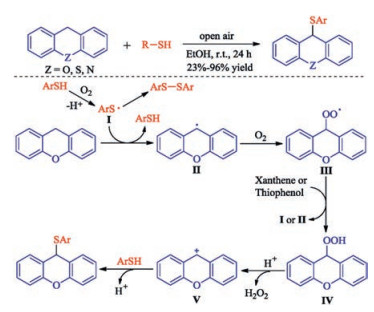
In 2019, a catalyst-free direct C(sp3)-H sulfenylation was achieved by Chen and co-workers via the autoxidative cross-dehydrogenative coupling (CDC) between xanthene derivatives and thiophenols in ethanol using air as the sole oxidant at room temperature [32]. This is the first report on the autoxidative cross-dehydrogenative coupling (CDC) between xanthene derivatives and thiophenols. This simple and efficient method provides a straightforward access to 9-sulfenylated xanthenes, 9, 10-dihydroacridines and thioxanthene with good functional group compatibility.
A plausible reaction mechanism for this transformation is outlined in Scheme 17. Firstly, thiyl radical Ⅰ and a proton are generated from the autoxidation of thiophenol. Then, 9-xanthenyl radical Ⅱ might be formed via a hydrogen abstraction from xanthene by radical Ⅰ. Thus, in the presence of oxygen, the oxidation of xanthene proceeds via radicals Ⅱ and Ⅲ to hydroperoxide Ⅳ, which generates H2O2 and a stabilized carbocation Ⅴ. Finally, thiophenol attacks benzylic cation Ⅴ, leading to the formation of desired product (Scheme 17).

|
Download:
|
| Scheme 17. Catalyst-free sulfenylation of xanthene derivatives. | |
{kind=link}
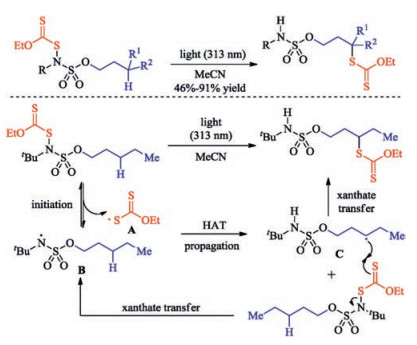
Moreover, Roizen group achieved the sulfamate ester guided selective sulfenylation of alkanes via light-initiation [33]. The research confirms the recently disclosed basic scientific insight that N-functionalized sulfamate esters can guide position-selective C(sp3)-H functionalization such as sulfenylation, azidation, allylation. Theoretically, this xanthate-transfer process could occur through a closed-cycle and/or a radical chain propagation mechanism. In these processes, light initiates N-S bond homolysis to convert N-xanthate into a xanthate radical and sulfamyl radical A. This sulfamyl radical is poised for an intramolecular C-H abstraction through a seven membered transition state to produce carbon-centered radical C, imparting position selectivity to this transformation. Then in a chain propagation mechanism, alkyl radical C reacts with another substrate molecule to furnish alkyl xanthate and another 1 equiv. of xanthate radical, which would then propagate this chain reaction (Scheme 18).

|
Download:
|
| Scheme 18. Light-initiated sulfenylation of alkanes. | |
{kind=link}
3. Enantioselective sulfenylation of C(sp3)H 3.1. N-(Thio)succinimides as sulfenylation reagents
The α-sulfenylation of ketones has been shown to generate an asymmetric quaternary carbon center, hence there is scope for developing enantioselective sulfenylated methods. In recent years, there are more and more routes to enantioselectively synthesize α-sulfenylated ketones and derivatives to be developed. Among these tactics, N-(thio)succinimides are the most commonly applied sulfenylated reagents, and most of the reaction substrates are concentrated in oxindoles and other heterocyclones.
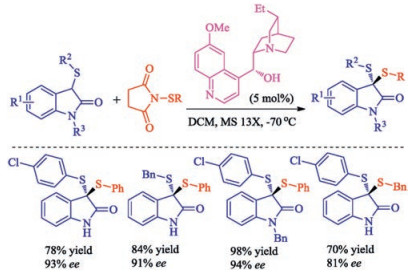
In 2015, Zhou group reported the first example of highly enantioselective synthesis of structurally diverse chiral dithioketals via asymmetric sulfenylation [34]. They speculated that cooperation between an H-bond donor and a Brønsted base might be helpful for this process, so when cinchona alkaloid derivative dihydroquinine was used as catalyst, structurally diverse chiral dithioketals could be synthesized smoothly with high enantioselectivity along with the broad substrate scope (Scheme 19).

|
Download:
|
| Scheme 19. Catalytic asymmetric sulfenylation to synthesize dithioketals. | |
{kind=link}
Using chiral bicyclic guanidine derivatives as catalysts, Liu and co-workers achieved highly enantioselective sulfenylation of oxindoles with N-(thio)succinimides [35]. The reactions worked smoothly in the presence of a chiral bicyclic guanidine and afforded the unprecedented sulfenylated products with good to excellent yields and enantioselectivities (up to 98%) (Scheme 20).

|
Download:
|
| Scheme 20. Chiralbicyclic guanidine catalyzed asymmetric sulfenylation of oxindoles. | |
{kind=link}
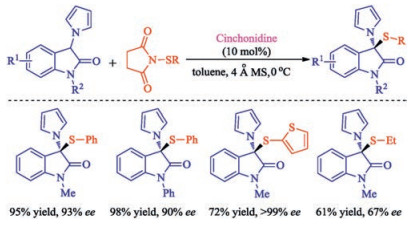
Organocatalytic asymmetric sulfenylation for the synthesis of 3, 3-disubstituted oxindoles bearing two different heteroatoms at the C3 position with commercially available cinchonidine as catalyst was established by Yuan group [36]. A wide range of optically active 3-thio-3-pyrrolyl-oxindoles could be smoothly obtained under mild conditions with satisfactory results (Scheme 21). In particular, this methods also was employed for enantioselective synthesis of selenium-containing oxindoles for the first time.

|
Download:
|
| Scheme 21. Cinchonidine catalyzed asymmetric sulfenylation of oxindoles. | |
{kind=link}
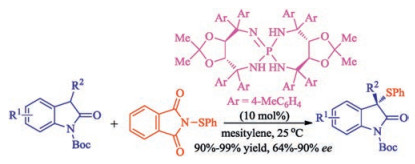
In 2016, Wang and co-workers achieved catalytic asymmetric sulfenylation of 3-substituted oxindoles through efficient catalysis by tartaric acid derived chiral iminophosphoranes with N-(phenylthio)phthalimide as the sulfur source [37]. A wide range of optically active 3-phenylthiooxindoles were obtained in excellent yields (90%–99%) and good enantiomeric excess (up to 90% ee) at room temperature (Scheme 22).

|
Download:
|
| Scheme 22. Chiral iminophosphoranes catalyzed asymmetric sulfenylation of oxindoles. | |
{kind=link}
An asymmetric sulfenylation of 3-CF3-oxindoles catalyzed by a cinchona alkaloid derivative was described by Xu and co-workers [38]. 3-CF3-oxindoles with electro-donating groups led to corresponding products in good yield and with good enantioselectivity using quinidine-OiPr as a catalyst (Scheme 23). Unfortunately, 3-CF3-oxindoles bearing electro-withdrawing groups were not applicable due to its significant decomposition at the optimal reaction conditions.

|
Download:
|
| Scheme 23. Quinidine-OiPr catalyzed asymmetric sulfenylation of oxindoles. | |
{kind=link}
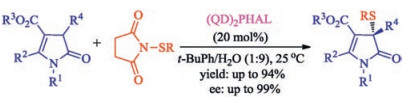
Except for oxindoles, other ketone and heterocyclones such as unsaturated butyrolactam [39], pyrazolones [40], benzofuran-2-ones [34, 35], β-ketocarbonyls [41] also can be asymmetrically sulfenylated by N-(thio)succinimides using chiral catalyst. For example, Mukherjee group described the first catalytic enantioselective α-sulfenylation of deconjugated butyrolactams using dimeric cinchona alkaloids as the catalyst in water-enriched reaction medium in 2017 [39]. This protocol delivers highly substituted and densely functionalized γ-lactams in moderate to high yields and generally with high enantioselectivities (Scheme 24). Interestingly, substantial rate acceleration was observed in water-enriched or aqueous media, possibly due to hydrophobic hydration effect.

|
Download:
|
| Scheme 24. Asymmetric sulfenylation of deconjugated butyrolactams. | |
{kind=link}
Using chiral iminophosphorane as the organocatalyst and pentane as the solvent under continuum solvation conditions, an excellent asymmetric sulfenylation of 4-substituted pyrazolones was developed by Han and co-workers [40]. In particular, this catalytic process offered the desired sulfenylated products in excellent yields with high levels of enantiomeric excess and features high efficiency, easy separation of products, low catalytic loadings and scale-up to grams without loss of enantioselectivity (Scheme 25).

|
Download:
|
| Scheme 25. Asymmetric sulfenylation of pyrazolones. | |
{kind=link}
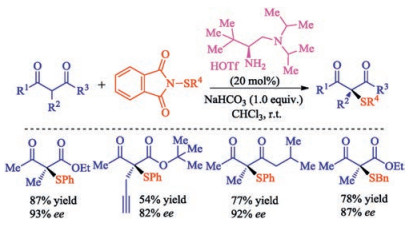
The asymmetric sulfenylation of acyclic β-ketocarbonyls is relatively few compared to (hetero)cyclones. In 2018, Mi and co-workers developed a direct asymmetric α-sulfenylation of β-ketocarbonyls using chiral primary amine as catalyst with good yields and excellent enantioselectivities [41]. The reaction proceeded via enamine catalysis with good stereocontrol for both cyclic and acyclic β-ketocarbonyls (Scheme 26).

|
Download:
|
| Scheme 26. Asymmetric sulfenylation of acyclic β-ketocarbonyls. | |
{kind=link}
3.2. Other sulfenylating reagents
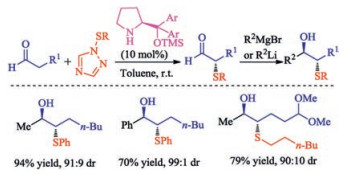
In addition to the most usually sulfur reagents N-(thio)succinimides, disulfides were also employed for the asymmetric sulfenylation of oxindoles in the presence of chiral phase-transfer catalyst derived from cinchona alkaloids by Itoh group [42]. The reaction was suggested to proceed in a radical mechanism. Therefore, this catalytic process only provides moderate enantioselectivities (up to 72%). In 2016, Sheppard and co-workers prepared the enantioenriched secondary alcohols via organocatalytic sulfenylation of aldehydes followed by organometallic addition and desulfurization [43]. Importantly, to provide enantioenriched desired α-phenylsulfenyl aldehydes in high enantiopurity through asymmetric organocatalytic sulfenylation is vital for this process (Scheme 27). This approach can provide access to enantiomerically enriched chiral building blocks which are difficult to access via existing approaches.

|
Download:
|
| Scheme 27. Asymmetric organocatalytic sulfenylation of aldehydes. | |
{kind=link}
4. Summary and prospect
Herein we present an overview of the recent five-year advances in the direct sulfenylation of C(sp3)-H for C-S bond formation under metal-free conditions. Various sulfenylating reagents such as thiols, disulfides, sodium sulfinates, sulfonyl chlorides, have been developed and employed for direct sulfenylation of C(sp3)-H bonds. In terms of eco-friend and atom economy, disulfides might be more ideal sulfur reagents than others. Although considerable efforts have been devoted and great advances also have been achieved, there are still lots of research that worth to be achieved. Direct sulfenylation of C-H bond is still a vigorous research field. Firstly, there are few research on inert α-H of alkanes which is a prospective virgin research field. Moreover, only a few reactions have been asymmetrically catalyzed and most on asymmetric sulfenylation of active α-H. In addition, there is no report about the asymmetric sulfenylation of inert α-H of alkanes. All of these need to be further explored and the results need to be further improved upon. Finally, the mechanisms of these reactions need to be further explored. More specific reaction mechanisms should help us to design and achieve more efficient and eco-friendly sulfenylations, and even with high enantioselectivity. There are opportunities and challenges coexisting in this field. We believe that more and more attention will be attracted to this area, and more innovative achievements can be presented in the near future.
AcknowledgmentWe are grateful for financial support from the Young Scholars Research Fund of Yantai University (No. HY19B06).
| [1] |
(a) R.J. Cremlyn, An Introduction to Organosulfur Chemistry, Wiley, New York, 1996; (b) D. Meng, W. Chen, W. Zhao, J. Nat. Prod. 70 (2007) 824-829; (c) M. Kvasnika, M. Urban, N.J. Dickinson, J. Sarek, Nat. Prod. Rep. 32 (2015) 1303-1330. |
| [2] |
M.H. Feng, B.Q. Tang, H.L. Steven, X.F. Jiang, Curr. Top. Med. Chem. 16 (2016) 1200-1216. DOI:10.2174/1568026615666150915111741 |
| [3] |
(a) D.A. Boyd, Angew. Chem. Int. Ed. 55 (2016) 15486-15502; (b) D. Wu, W. Pisula, M.C. Haberecht, X. Feng, K. Müllen, Org. Lett. 11 (2009) 5686-5689; (c) S.M. Yang, J.J. Shie, J.M. Fang, S.K. Nandy, Y.Y. Chang, J. Org. Chem. 67 (2002) 52085215. |
| [4] |
(a) J.C. Carretero, Chem. Commun. 47(2011) 2207-2211; (b) H. Pellisier, Chiral Sulfur Ligands in Asymmetric Catalysis, RSC Catalysis Series 2, Cambridge, 2009. |
| [5] |
(a) A. Kausar, S. Zulfiqar, M.I. Sarwar, Pol. Rev. 54 (2014) 185-267; (b) A.S. Rahate, K.R. Nemade, S.A. Waghuley, Rev. Chem. Eng. 29 (2013) 471-489; (c) N. Spassky, Phosphorus Sulfur Silicon Relat. Elem. 74 (1993) 71-92. |
| [6] |
(a) J.F. Hartwig, Nature 455 (2008) 314-322; (b) Q. Lu, J. Zhang, F.L. Wei, et al., Angew. Chem. Int. Ed. 52 (2013) 7156-7159; (c) Q.Q. Lu, J. Zhang, G.L. Zhao, et al., J. Am. Chem. Soc.135 (2013) 11481-11484; (d) S.H. Hao, L.X. Li, D.Q. Dong, Z.L. Wang, Chin. J. Catal. 38 (2017) 1664-1667; (e) L.H. Lu, S.J. Zhou, W.B. He, et al., Org. Biomol. Chem. 16 (2018) 9064-9068; (f) L.Y. Xie, Y.J. Li, J. Qu, et al., Green Chem. 19 (2017) 5642-5646; (g) F.L. Zeng, X.L. Chen, S.Q. He, et al., Org. Chem. Front. 6 (2019) 1476-1480; (h) D. Yang, P. Sun, W. Wei, et al., Chem. -Eur. J. 24 (2018) 4423-4427; (i) L. Penga, Z. Hua, Z. Tang, Y. Jiao, X. Xu, Chin. Chem. Lett. 30 (2019) 1481-1487. |
| [7] |
(a) M. Martinek, M. Korf, J. Srogl, Chem. Commun. 46 (2010) 4387-4389; (b) S.K. Sahoo, A. Banerjee, S. Chakraborty, B.K. Patel, ACS Catal. 2 (2012) 544-551; (c) O. Saidi, J. Marafie, A.E. Ledger, et al., J. Am. Chem. Soc. 133 (2011) 19298-19301; (d) N. Umierski, G. Manolikakes, Org. Lett. 15 (2013) 4972-4975; (e) Z. Wu, H. Song, X. Cui, et al., Org. Lett. 15 (2013) 1270-1273; (f) B. Niu, L. Xu, P. Xie, et al., ACS Comb. Sci. 16 (2014) 454-458. |
| [8] |
(a) S.N. Zhang, S.H. Yang, L.H. Huang, et al., Chin. J. Org. Chem. 35 (2015) 2259-2274; (b) R. Chitrakar, A. Subbarayappa, Chem. Rec. 17 (2017) 1; (c) Y.Y. Liu, J. Xiong, L. Wei, Chin. J. Org. Chem. 37 (2017) 1667-1680; (d) D.Q. Dong, S.H. Hao, D.S. Yang, L.X. Li, Z.L. Wang, Eur. J. Org. Chem. 2017 (2017) 6576-6592; (e) L. Li, Y.Q. Ding, Mini-Rev. Org. Chem. 14 (2017) 407-418; (f) R. Dalpozzo, Org. Chem. Front. 4 (2017) 2063-2078; (g) M. Freckleton, A. Baeza, L. Benavent, R. Chinchilla, Asian. J. Org. Chem. 7 (2018) 1006-1014; (h) C.A. Jin, Q. Xu, G.F. Feng, Y. Jin, L.Y. Zahng, Chin. J. Org. Chem. 38 (2018) 775-790. |
| [9] |
(a) A. Ghaderi, Tetrahedron 72 (2016) 4758-4782; (b) K.L. Dunbar, D.H. Scharf, A. Litomska, C. Hertweck, Chem. Rev. 117 (2017) 5521-5577; (c) J. Zhu, W.C. Yang, X.D. Wang, L. Wu, Adv. Synth. Catal. 360 (2018) 386-400; (d) Y. Luo, Y. Ma, Z. Hou, J. Am. Chem. Soc. 140 (2018) 114-117. |
| [10] |
B.V. Varun, K. Gadde, K.R. Prabhu, Org. Lett. 17 (2015) 2944-2947. DOI:10.1021/acs.orglett.5b01221 |
| [11] |
H. Cao, J. Yuan, C. Liu, X.Q. Hu, A.W. Lei, RSC Adv. 5 (2015) 41493-41496. DOI:10.1039/C5RA04906G |
| [12] |
Y. Jiang, J.X. Zou, L.T. Huang, et al., Org. Biomol. Chem. 16 (2018) 1641-1645. DOI:10.1039/C8OB00080H |
| [13] |
Q. Chen, X. Wang, C. Wen, et al., RSC Adv. 7 (2017) 39758-39761. DOI:10.1039/C7RA06904A |
| [14] |
Y. Liu, S.S. Badsara, Y. Liu, C. Lee, RSC Adv. 5 (2015) 44299-44305. DOI:10.1039/C5RA07204B |
| [15] |
R. Rahaman, N. Devi, P. Barman, Tetrahedron Lett. 56 (2015) 4224-4227. DOI:10.1016/j.tetlet.2015.05.062 |
| [16] |
N. Devi, R. Rahaman, K. Sarma, P. Barman, Eur. J. Org. Chem. 2016 (2016) 384-388. DOI:10.1002/ejoc.201501148 |
| [17] |
B.M. Trost, Chem. Rev. 78 (1978) 363-382. DOI:10.1021/cr60314a002 |
| [18] |
B. Hu, Q. Zhang, S. Zhao, et al., Adv. Synth. Catal. 361 (2019) 49-54. DOI:10.1002/adsc.201801138 |
| [19] |
Y. Siddaraju, K.R. Prabhu, Org. Lett. 18 (2016) 6090-6093. DOI:10.1021/acs.orglett.6b03084 |
| [20] |
Y. Siddaraju, K.R. Prabhu, J. Org. Chem. 83 (2018) 2986-2992. DOI:10.1021/acs.joc.7b03290 |
| [21] |
Y. Siddaraju, K.R. Prabhu, Org. Biomol. Chem. 15 (2017) 5191-5196. DOI:10.1039/C7OB00561J |
| [22] |
N. Devi, R. Rahaman, K. Sarma, T. Khan, P. Barman, Eur. J. Org. Chem. 2017 (2017) 1520-1525. |
| [23] |
(a) P.N. Kalaria, S.P. Satasia, J.R. Avalani, D.K. Raval, Eur. J. Med. Chem. 83 (2014) 655-664; (b) S.C. Karad, V.B. Purohit, D.K. Raval, Eur. J. Med. Chem. 84 (2014) 51-58. |
| [24] |
R.D. Kamani, V.B. Purohit, R.P. Thummar, et al., ChemistrySelect 2 (2017) 9670-9673. DOI:10.1002/slct.201701924 |
| [25] |
(a) H. Jin, W. Wang, Z. Yang, et al., Heterocycles 96 (2018) 1786-1794; (b) X. Zhao, X. Lu, A. Wei, et al., Tetrahedron Lett. 57 (2016) 5330-5333; (c) X. Zhao, A. Wei, X. Lu, K. Lu, Molecules 22 (2017) 1208-1219. |
| [26] |
X. Liu, H. Cui, D. Yang, et al., RSC Adv. 6 (2016) 51830-51833. DOI:10.1039/C6RA09739A |
| [27] |
Q. Chen, Y. Huang, X. Wang, et al., Tetrahedron Lett. 58 (2017) 3928-3931. DOI:10.1016/j.tetlet.2017.08.067 |
| [28] |
(a) A.F. Vaquer, A. Frongia, F. Secci, E. Tuveri, RSC Adv. 5 (2015) 96695-96704; (b) H.W. Noh, C. Lee, H.Y. Jang, Bull. Korean Chem. Soc. 38 (2017) 389-391; (c) J.Q. Zhao, S.W. Luo, X.M. Zhang, et al., Tetrahedron 73 (2017) 5444-5450. |
| [29] |
Y. Li, F. Zhu, Z. Wang, X.F. Wu, Chem. -Asian J. 11 (2016) 3503-3507. DOI:10.1002/asia.201601376 |
| [30] |
D. Wang, Z. Liu, Z. Wang, X. Ma, P. Yu, Green Chem. 21 (2019) 157-163. DOI:10.1039/C8GC03072C |
| [31] |
Y. Liu, X. Yuan, K. Su, Y. Tian, B. Chen, Eur. J. Org. Chem. 2019 (2019) 1649-1652. DOI:10.1002/ejoc.201801806 |
| [32] |
Q. Chen, G. Yu, X. Wang, Y. Ou, Y. Huo, Green Chem. 21 (2019) 798-802. DOI:10.1039/C8GC03898H |
| [33] |
S.K. Ayer, J.L. Roizen, J. Org. Chem. 84 (2019) 3508-3523. DOI:10.1021/acs.joc.9b00105 |
| [34] |
K. Liao, F. Zhou, J. Yu, W. Gao, J. Zhou, Chem. Commun. 51 (2015) 16255-16258. DOI:10.1039/C5CC07010D |
| [35] |
L. Huang, J. Li, Y. Zhao, et al., J. Org. Chem. 80 (2015) 8933-8941. DOI:10.1021/acs.joc.5b01606 |
| [36] |
Y. You, Z. Wu, Z. Wang, et al., J. Org. Chem. 80 (2015) 8470-8477. DOI:10.1021/acs.joc.5b01491 |
| [37] |
X. Gao, J. Han, L. Wang, Synthesis 48 (2016) 2603-2611. DOI:10.1055/s-0035-1560435 |
| [38] |
Y. E, T. Yuan, L. Yin, Y. Xu, Tetrahedron Lett. 58 (2017) 2521-2524. DOI:10.1016/j.tetlet.2017.05.015 |
| [39] |
S.J. Singha Roy, S. Mukherjee, Org. Biomol. Chem. 15 (2017) 6921-6925. DOI:10.1039/C7OB01714F |
| [40] |
J. Han, Y. Zhang, X.Y. Wu, H.N.C. Wong, Chem. Commun. 55 (2019) 397-400. DOI:10.1039/C8CC09049A |
| [41] |
L. Cui, Y. You, X. Mi, S. Luo, Org. Chem. Front. 5 (2018) 2313-2316. DOI:10.1039/C8QO00496J |
| [42] |
K. Nagata, D. Sano, O. Aoyama, et al., Heterocycles 92 (2016) 631-635. DOI:10.3987/COM-16-13414 |
| [43] |
F. Rota, L. Benhamou, T.D. Sheppard, Synlett 27 (2016) 33-36. DOI:10.1055/s-0035-1560769 |