 2020, Vol. 31
2020, Vol. 31 

b Research Institute of Sun Yat-Sen University in Shenzhen, Shenzhen 518057, China
Hydrogel is a kind of polymer network system, which is soft in nature and could maintain a certain shape while absorbing large amounts of water [1, 2]. Owing to the viscoelastic properties similar to living tissues and the permeability to various types of molecules, hydrogels have received extensive attentions in the fields of biomedicine, biochemistry and so on [3-10]. According to the responsibility to external stimuli, hydrogels can be divided into traditional hydrogels and intelligent hydrogels. Traditional hydrogels are insensitive to environmental changes, while intelligent hydrogels, also known as stimuli-responsive hydrogels, have the ability to respond to external stimuli, such as temperature [11-15], pH [16-19], light [20-23], magnetism [24, 25], ion concentration [26], electrical field [27], solvent composition [28, 29], pressure [30], biomolecules [31-33]. When external stimulus is applied, the network of intelligent hydrogel will shrink or swell accordingly, resulting in significant changes of the hydrogel matrix properties [34, 35]. Due to the remarkable responsiveness, intelligent hydrogels have been widely utilized in various fields, such as controlled release drug carriers [36-40], tissue engineering [41], chemical/biological separators [42, 43], gene carriers [44], optical microlens [45]. Although intelligent hydrogels show bright application prospects, some serious problems remain to be solved, such as how to achieve a controllable stimulus response and improve the response sensitivity.
It is well known that the chemical composition and molecular structure of the polymer precursors act a pivotal role in determining the hydrogel properties. In recent years, numerous efforts have been made to overcome the problems faced by intelligent hydrogels through regulating the structure of polymer precursors [46-48]. Among them, due to the advantages of wide range of suitable monomers and strong molecular design ability, reversible addition-fragmentation chain transfer (RAFT) polymerization, one of the controlled/living radical polymerizations (CRP), has developed into a strong tool to design the structure of polymer precursors [49-51]. RAFT polymerization can easily achieve the fabrication of block, graft and star copolymers with narrow molecular weight distribution, which providing a versatile platform for the manufacture and performance upgrading of intelligent hydrogels [52-54]. Nowadays, a large number of literatures have reported on the intelligent hydrogels based on RAFT polymerization, which exhibit a controllable stimulus response or a high response sensitivity [55, 56]. It is well worth summarizing the progress and future development of intelligent hydrogels based on RAFT polymerization. However, as far as we know, there is little literature to do such work.
The aim of this review is to sum up the recent progresses of intelligent hydrogels based on RAFT polymerization. In the present article, RAFT polymerization will be introduced in detail first, followed by the construction of single response and multiresponse hydrogels based on RAFT polymerization, respectively, and their applications in drug-controlled release. At the end of this article, current challenges and future prospect for RAFT based intelligent hydrogels will be discussed.
2. RAFT polymerization 2.1. Controlled/living radical polymerizations (CRP)Since the concept was first put forward in 1956 [57], living polymerization has gradually developed into a research focus with industrial application value and academic significance in the field of polymer chemistry [58, 59]. Living polymerization refers to a polymerization without both chain transfer reactions and chain termination, and the active species can still remain active even after the monomers are all consumed [60-62]. Based on these characteristics, living polymerization makes the molecular design of polymer chains a reality.
According to the initiation mechanism, living polymerization can be divided into living radical polymerization, living anionic polymerization, living cationic polymerization, living coordination polymerization and so on [63-66]. Compared with the other living polymerizations, living radical polymerization are suitable for more kinds of monomers and can be processed under relatively mild conditions [67, 68]. Furthermore, Matyjaszewski et al. defined the concept of controlled/living radical polymerization (CRP), in which the growth chain radicals can reversibly form dormant active species with other substances and have the potential to continue to grow with narrow molecular weight distribution (PDI < 1.3) [69]. At present, the most influential and mature strategies of CRP including stable free radical polymerization (SFRP), initiation-transfer-terminator (Iniferter), atom transfer radical polymerization (ATRP), reversible addition-fragmentation chain transfer (RAFT), and so on [70-74]. Among them, RAFT polymerization has attracted numerous attentions due to its ability to synthesize various block copolymers with narrow molecular weight distribution, wide range of reaction temperature (generally from 40 ℃ to 160 ℃), and reaction process without protecting groups.
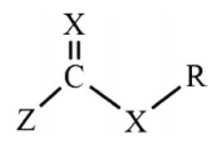
2.2. Mechanism of RAFT polymerizationIn general, organic compounds with large chain transfer constants used in RAFT polymerization are called RAFT agents, as shown in Fig. 1. RAFT agents are mainly divided into two categories with methylene or sulfur as X group, respectively [75, 76]. However, RAFT agents with methylene as X group were not commonly used as their synthesis and post-treatment are troublesome [77]. The most commonly used RAFT agents with sulfur as X group including trithioesters and dithioesters, such as trithiocarbonates, xanthates, dithiocarboxylic acid esters, dithiocarbamate [78-83]. The Z group can activate C=S bond so that it can react with free radicals. The R group can be easily cleaved, and the resulted free radicals can effectively re-initiate the polymerization of the monomers. By modulating the structure of R and Z groups, the corresponding chain transfer constant can be changed by 5 orders of magnitude.

|
Download:
|
| Fig. 1. Schematic representation of RAFT agent. | |
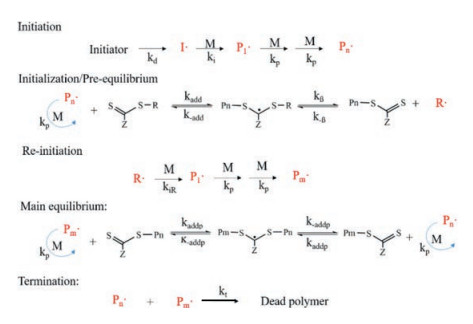
A typical RAFT polymerization could be divided into three phases, initiation phase, chain transfer phase and termination phase. In the initiation phase, monomer (M) is initiated by initiator (I) to form free radicals (Pn·). In the chain transfer phase, Pn· reacts with the chain transfer agent (RA) to form PnA (·) R, which can be reversibly broken into new chain transfers PnA and free radical R·. R· can reinitiate M to obtain Pm· and PmA, possessing the same chain transfer properties as the original RA. As the rate of addition or fragmentation is much faster than that of chain growth, RAFT agent rapidly switches between active and dormant free radicals, leading to the growing polymer chain with narrow molecular weight distribution (Fig. 2) [49]. Such mechanism endows RAFT polymerization the ability to fabricate desirable block copolymers with pre-designed composition, block sequence and molecular weight by controlling the feeding sequence and amount of preselected monomers in polymerization. The ability to achieve the multidimensional regulation of polymer chain structure makes RAFT polymerization a fantastic platform for the preparation of intelligent hydrogels with improved response performance.

|
Download:
|
| Fig. 2. Equilibria of RAFT polymerization. | |
2.3. Strategy for the fabrication of intelligent hydrogels based on RAFT polymerization
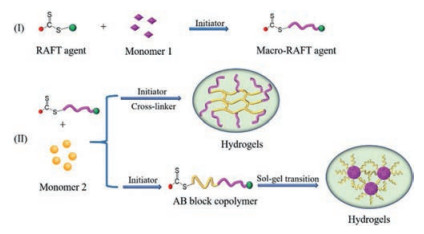
Owing to the unique polymerization mechanism, RAFT polymerization can efficiently achieve the fabrication of hydrogel precursors with well-defined structure, such as block copolymer, graft copolymer, star copolymer, which provides a huge library of building materials for the construction of intelligent hydrogels [84-86]. In addition, RAFT agents can be modified with a variety of functional groups, further enriching the construction of intelligent hydrogels [87, 88]. At present, the most commonly utilized strategies for intelligent hydrogel preparation include one-step RAFT polymerization, sequential RAFT polymerization and the combination of RAFT polymerization and click chemistry.
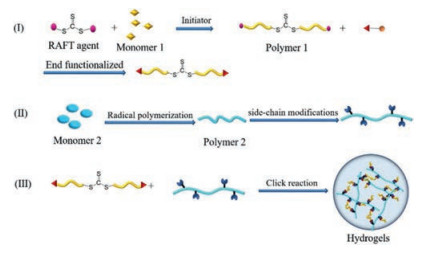
For one-step polymerization, monomers and cross-linkers are added at once, then react under the modulating of RAFT agent, which usually applied for the preparation of single response hydrogel with uniform structure (Fig. 3). Sequential RAFT polymerization, in which different monomers are polymerized sequentially, is frequently employed to synthesize AB, ABA and ABC copolymers for the preparation of comb-type intelligent hydrogels or multiresponse hydrogels (Fig. 4, Fig. 5, Fig. 6). Under certain conditions, some kinds of RAFT agents can be end-functionalized for click chemistry, which provides a new design idea for the construction of intelligent hydrogels by the combination of RAFT polymerization and click chemistry (Fig. 7). The advantages and limitations of these polymerization strategies are summarized in Table 1.

|
Download:
|
| Fig. 3. Schematic illustration of one-step RAFT polymerization. | |

|
Download:
|
| Fig. 4. Schematic representation of AB copolymers for the preparation of intelligent hydrogels by sequential RAFT polymerization. | |

|
Download:
|
| Fig. 5. Schematic illustration of ABA copolymers for the preparation of intelligent hydrogels by sequential RAFT polymerization. | |

|
Download:
|
| Fig. 6. Schematic illustration of ABC copolymers for the preparation of intelligent hydrogels by sequential RAFT polymerization. | |

|
Download:
|
| Fig. 7. Schematic illustration of the combination of RAFT polymerization and click chemistry. | |
|
|
Table 1 The advantages and limitations of three kinds of polymerization strategies. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
3. Single responsive hydrogels based on RAFT polymerization 3.1. Temperature–responsive hydrogels based on RAFT polymerization
At present, temperature-responsive hydrogels, also known as thermo-sensitive hydrogels, which are sensitive to ambient temperature, have developed into one of the most studied intelligent hydrogels [89, 90]. In general, the swelling behavior of thermo-sensitive hydrogels does not change gently, but shrink or swell sharply at a certain temperature, known as critical phase transition temperature (Tc) [91]. According to the temperature response behavior, thermo-sensitive hydrogels can be divided into two categories: thermo-expansion hydrogels and thermo-shrink hydrogels [92, 93]. Nowadays, hydrogels based on poly(N-isopropylacrylamide) (PNIPAAm), a kind of thermo-shrink hydrogels, have attracted the most attentions in the thermo-sensitive hydrogels field [94-96]. As PNIPAAm performs a lower critical solution temperature (LCST) at about 32 ℃ [97], which is very close to human physiological temperature (37 ℃), PNIPAAm-based hydrogels have been widely utilized for biomedical application. However, traditional PNIPAAm-based hydrogels generally possessed low response rate and monotonous performance, which severely limited their application prospects [98-101]. Recently, the sequential RAFT polymerization, in which the functional molecular chains can be introduced into the polymer network in a graft or block copolymerization manner by the selection of the RAFT agent, has developing into an efficient tool to prepare PNIPAAm-based hydrogels with improved performance.
3.1.1. PNIPAAm-based hydrogels with rapid response behaviorsNumerous studies have shown that the introduction of hydrophilic polymer chains in the hydrogel network can accelerate the flow of water molecules in the matrix, thereby increasing the response rate of the hydrogel, which is beneficial to improve the sensitivity of drug release [102]. Based on such idea, Zheng's group has made a lot of efforts to fabricate PNIPAAm-based hydrogel with rapid response behaviors by sequential RAFT polymerization.
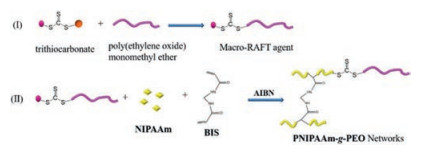
For example, Zheng et al. prepared PNIPAAm-based hydrogel under the modulating of poly(ethylene oxide)-terminated trithiocarbonate as macro-RAFT agent (Fig. 8) [103]. After such processing, PEO chains were efficiently grafted onto PNIPAAm network. The swelling test indicated that the obtained PNIPAAm-g-PEO hydrogels exhibited faster responses to external temperature changes compared with the control PNIPAAm hydrogel, owing to the hydrophilicity of the grafted hydrophilic PEG chain. Next, Zheng et al. used 1, 4-phenylenebis(methylene)bis(ethyl xanthate) as RAFT agent to synthesize α, ω-dixanthate-terminated poly(N-vinyl pyrrolidone) (PVPy), which work as macro-RAFT agent to fabricate PVPy-b-PNIPAAm hydrogel [104]. The swelling tests revealed that the inclusion of PVPy nanophases accelerated the diffusion of water molecules, leading to the faster response rate of PNIPAAm-b-PVPy hydrogels than the control PNIPAAm hydrogel. Similarly, Zheng et al. prepared PNIPAAm-b-PAA hydrogel with α, ω-dicarbonotrithioate-terminated poly(acrylic acid) as macro-RAFT agent by sequential RAFT polymerization [105]. Although the swelling ratios of PNIPAAm-b-PAA hydrogel decreased sharply, the response rate increased significantly, as the result of the formation of stable interchain complexation between PNIPAAm and PAA chains. Furthermore, Zheng et al. synthesized PNIPAAm-b-PSSNa hydrogel via sequential RAFT polymerization with α, ω-didithiobenzoate-terminated PSSNa as macro-RAFT agent. Due to the formation of PSSNa microdomains, which accelerated the flow of water molecules within hydrogel matrix, the thermo-responsive properties of the nanostructured hydrogels were improved significantly [106].

|
Download:
|
| Fig. 8. Schematic illustration of the formation of PNIPAAm-g-PEO hydrogels. | |
{kind=link}
3.1.2. PNIPAAm-based hydrogels as drug-controlled release systems
Owing to the LCST approaching to human body temperature, PNIPAAm-based hydrogels have significant potential for biomedical applications [107, 108], especially as drug-controlled release systems. However, the performance of traditional PNIPAAm-based hydrogels was too monotonous to meet practical application needs. Recently, RAFT polymerization has suggested new avenues for the preparation of high-performance PNIPAAm-based hydrogels for drug release applications, due to its ability to achieve the efficient and diverse introduction of functional molecular chains into the hydrogel matrix.
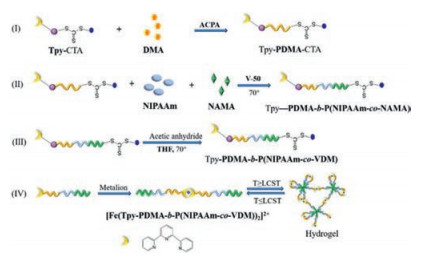
Numerous researches have been reported RAFT-generated thermo-responsive copolymers of PNIPAAm acted as drug carriers. For example, Bohec et al. fabricated a well-defined Tpy-PDMA-b-P(NIPAAm-co-VDM) hydrophilic block copolymer by sequential RAFT polymerization with terpyridine (Tpy-CTA) as RAFT agent (Fig. 9) [109]. A metallic bis-terpyridine complex (Fe(Tpy-PDMA-b-P(NIPAAm-co-VDM))2]2+) could be formed by the introduction of Fe2+ into a concentrated solution of Tpy-PDMA-b-P(NIPAAm-co-VDM) (8% w/v). With the increasing of temperature, a hydrogel could be formed by the self-assembly of the thermo-responsive P(NIPAAm-co-VDM) blocks. Such sol-gel thermo-reversible transition favor Tpy-PDMA-b-P(NIPAAm-co-VDM) micelles the potential as thermo-responsive drug release systems. Zeng et al. synthesized poly(N-isopropylacrylamide)-g-poly(sulfobetaine methacrylate) hydrogel (GH) by sequential RAFT polymerization with 2-(2-carboxyethylsulfanylthiocarbonyl sulfanyl) propionic acid as RAFT agent [110]. Compared to traditional PNIPAAm-co-PSBMA hydrogel (CH), GH performed a faster shrinking rate (losing more than 72% of the water within 15min) owning to a highly porous architecture. The in vitro test showed that the sustained release of tetracycline hydrochloride from GH could reach 48h, which was much longer than that of CH (only 5h). Tang et al. synthesized thermo-sensitive di-block copolymer, PNIPAAm-b-POAG, in which POAG acted as a biocompatible segment, via sequential RAFT polymerization [111]. The excellent sol-gel transition performance within a temperature range, covered the physiological conditions, enabled PNIPAAm-b-POAG micelles to transform into hydrogel immediately after the injection into physiological conditions. Especially, the in vitro test indicated that such PNIPAAm-b-POAG hydrogel could realize the sustained release of methylene blue for 120h. Prosperiporta et al. synthesized copolymers with varying NIAAm, AAm, acrylic acid N-hydroxysuccinimide (NAS), and (R)-α-acryloyloxy-β, β-dimethyl-γ-butyrolactone (DBA) by RAFT polymerization with 2-(dodecylthio-carbonothioylthio)-2-methylpropionic acid as RAFT agent [112]. After the injection into physiological conditions, poly(NIPAAm-AAm-NAS-DBA) copolymers could undergo a sol-gel transition by the trigger of temperature and work as a scaffold for controlled release of ophthalmic therapeutics. The physical properties and drug release behavior of such scaffold could be precisely modulated by varying monomer ratio and molecular weight of each monomer.

|
Download:
|
| Fig. 9. Schematic illustration of the formation of Tpy-PDMA-b-P(NIPAAm-co-VDM) hydrogel. | |
{kind=link}
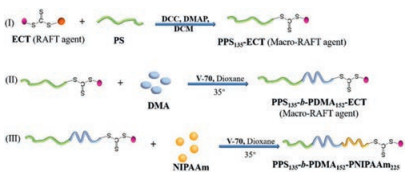
Duvall et al. [113] fabricated three kinds of ABC tri-block copolymers-based hydrogels with desirable degradability and drug release performance, PPS135-b-PDMA152-b-PNIPAAM225 (PDN), PCL85-b-PDMA150-b-PNIPAAM150 (CDN), and PLGA60-b-PDMA148-b-PNIPAAM152 (LGDN) (Fig. 10). In this work, hydrophilic PDMA and thermo-responsive PNIPAAm worked as the "B" and "C" blocks, respectively, while degradable materials with different degradation mechanisms, poly(propylene sulfide), poly(ε-caprolactone) and poly(D, L-lactide-co-glycolide) worked as the "A" blocks. Typically, hydroxyl end-functional PPS was synthesized based on anionic ring opening reaction, then coupled with the RAFT agent end functionalized by carboxyl to work as macro-RAFT agent. With the addition of DMA and NIPAAm in sequence, PDN was obtained. CDN and LGDN were also prepared by the similar method as described for PDN. The micelle solutions of these ABC tri-block copolymers could preload with therapy agents and immediately transformed into hydrogel at body temperature. The release profile of Nile Red indicated that the three kinds of hydrogels performed different release behaviors, fast release for LGDN, slow release for CDN and slow but more consistent release for PDN. Furthermore, the in vivo test showed that these hydrogels implants could improve robust cellular infiltration without any inflammatory response. Such a series of hydrogels with designable degradation mechanisms and drug release performance were also promising vehicles for the delivery of other therapy agent. Similarly, Li et al. [114] have synthesized hydrogels based on P(NIPAAm-FPA-DMA) copolymers, and the in vitro toxicity test demonstrated such hydrogels behaved good biocompatibility. Notably, for the in vitro release of Dox, such hydrogels enhanced drug release due to the high mobility of the polymer chains at high temperature.

|
Download:
|
| Fig. 10. Schematic representation of the formation of PDN hydrogel. | |
{kind=link}
Jiang et al. fabricated ABA tri-block copolymers with controllable MW and structure, in which the inner B block of hydrolysable betaine ester (HBE) was flanked by two outer A blocks of PNIPAAm, by sequential RAFT polymerization [115]. Similar to the other PNIPAAm-based ABA block copolymers, the micelles of PNIPAAm-PHBE-PNIPAAm copolymer solutions undergo a sol-gel transition upon physiological conditions. Besides, due to the positive surface charge, PHBE facilitated both the controlled release of antimicrobial drug with counter ion and the attachment of mammalian cells during tissue regeneration. Such hydrogel had promising applications as antimicrobial wound dressing.
3.2. pH-Responsive hydrogels based on RAFT polymerizationSince the first report by Tanaka in 1980 [116], pH-responsive hydrogels have attracted more and more attentions. The pH-responsive hydrogels generally contain alkaline or acidic groups (such as carboxyl, sulfonic or amino groups) that ionize as the pH changes [117-119], thus resulting in the dissociation of hydrogen bonds of polymer chains within networks and producing a discontinuous change in the swelling volume. The pH-responsive hydrogels prepared by traditional methods usually exhibited heterogeneous matrix properties and monotonous functions, which were not conducive for their applications. At present, RAFT polymerization has been frequently utilized to construct pH-responsive hydrogels due to its convenience in the preparation of polymer precursors with well-defined structures and the introduction of functional components.
Armes et al. constructed a novel hydrogel based on worm-like diblock copolymers, which were prepared by glycerol monomethacrylate (GMA) and 2-hydroxypropyl methacrylate (HPMA) with carboxylic acid-based RAFT agent [120]. By increasing the solution pH, the ionization of carboxylic acid end-group induced the sol-gel transition, leading to the de-gelation of GMA-HPMA hydrogel at once. The TEM and rheology tests indicated that only minimal amounts of carboxylic acid group could drive such reversible morphological transition. Li et al. fabricated a novel covalent dynamic hydrogel based on RAFT polymerization [121]. In this work, poly(2, 2-bis(hydroxymethyl)butylacrylate) (PHBA) and poly(VPB-co-DMA) with well-defined structure were first separately synthesized by RAFT polymerization. After the mixing of the two polymeric precursors, hydrogels could form rapidly, due to the reversible phenylborate ester bonds between phenylboronic acid and 1, 3-diol. Upon modulating solution pH, such hydrogel could transform back into polymer solutions. Besides, such hydrogel performed excellent self-healing behavior without any other treatment. The pH induced reversible sol-gel transition and self-healing ability enable the PHBA-poly(VPB-co-DMA) hydrogel to work as a promising drug carrier. Furthermore, Li et al. functionalized PHBA with single-walled carbon nanotubes (f-SWCNTs) by a nitrene addition reaction [122]. The obtained f-SWCNTs modified PHBA-poly(VPB-co-DMA) hydrogel exhibited remarkably improved mechanical performance, which was favor for practical applications. Kitiri et al. constructed a multi-functional double–network (DN) hydrogel comprised of a first pH-responsive network by sequential RAFT polymerization and a second mechanical enhanced network by free radical photopolymerization (Fig. 11) [123]. Such pH-responsive hydrogel with remarkable mechanical performance provided a reference for the next-generation biomaterials.

|
Download:
|
| Fig. 11. Schematic illustration of the formation of the multi-functional double-network (DN) hydrogel. | |
{kind=link}
Guo et al. synthesized well-defined copolymers based on diacetone acrylamide (DAAM) and acrylamide (AM) by one-step RAFT polymerization, then dynamically covalent bonded by hexanedihydrazide, resulting in the gelation [124]. The mechanical property and gelation concentration of this hydrogel could be modulated by varying the monomers ratio. Besides reversible sol–gel transition induced by pH alternation, such hydrogel exhibited self-healing ability without extra treatment. In addition, the in vitro test showed that this hydrogel possessed pH-responsive controlled release of rhodamine B. The rhodamine B burst released initially and subsequently sustained until over 36 h at pH 6.0, 76 h at pH 7.4 and 17 h at pH 2.5, respectively. The studies showed that the release of drug in a proper environment (pH 7.4) was more efficient than in an acidic environment, which might depend on the stability of hydrogel.
Liu et al. employed another strategy to prepare pH-responsive hydrogel with improved mechanical properties [125]. In this study, graphene/PAA hydrogel was synthesized by RAFT polymerization with graphene modified RAFT agent. Compared with traditional PAA hydrogel, graphene/PAA hydrogel exhibited more excellent elastic properties. Most importantly, the pH responsive hydrogel could be utilized as drug carrier. Compared to pH 2.0, the in vitro test showed that this hydrogel could realize more efficiently controlled release of doxorubicin in the intestine environment. The conclusion was similar to the research of Guo et al. Moreover, Wang et al. directly utilized the anticancer drug podophyllotoxin derivative hydrophobic monomer to synthesize the poly(triethylene glycol methacrylate)-b-poly(podophyllotoxin methacrylate) copolymers via RAFT polymerization [126]. The sustained release manner of podophyllotoxin was depended on pH value. In addition, Wang et al. prepared a pH-sensitive hydrogel that could degrade and release antibiotics when bacterial infection resulting in acidic environment [127]. Hydrogel was synthesized by copolymerization of vancomycin, PAA and PDMAEMA which was prepared via RAFT polymerization. Importantly, the hydrogel was demonstrated good biocompatibility.
3.3. Other single stimuli hydrogels based on RAFT polymerizationRecently, except for temperature and pH, other stimulus including light, magnetism, chemical agents and so on have also been commonly employed to trigger hydrogels for biomedical applications [128-131], such as drug delivery herein. RAFT polymerization provided a versatile platform to functionalize hydrogels with such response performances.
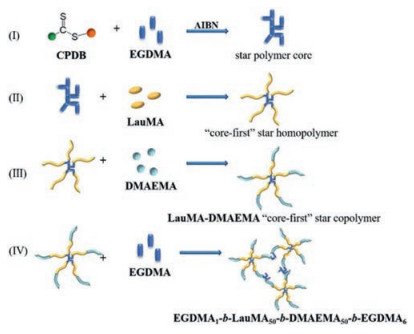
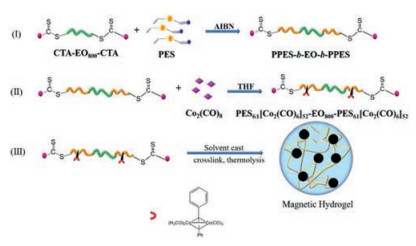
For example, Banerjee et al. synthesized a novel PCL based macro-RAFT agent by the combination of ring opening polymerization of ε-caprolactone and xanthate mediated RAFT polymerization [132]. The obtained macro-RAFT agent was employed to fabricate fluorescence responsive ionic block copolymers (BCPs), PCL-b-PMTAC and PCL-b-PSS, based on cationic PMTAC and anionic PSS with the copolymerization of a trace amount of fluorescein O-acrylate and 9-anthryl methylmethacrylate respectively to endow both BCPs fluorescent. With the cooperation of the two oppositely charged BCPs, a fluorescence active polyacrylamide-based hydrogel with self-healing ability was obtained. In addition, MTT assay experiments indicated that such hydrogel was non-toxic, which was favor for the application in the biomedical applications. Jiang et al. employed PEO based macro-RAFT agent to construct a novel ABA triblock copolymer (Fig. 12) with well-defined structure based on 4-(phenylethynyl)styrene (PES) [133]. By the reaction between alkyne-functionalized PPES block and Co2(CO)8, triblock copolymer/cobalt adducts were obtained. When subjected to 120 ℃, a magnetic hydrogel could be formed due to the hydrophobic PPES domains. Swelling tests, actuation tests and rheological studies showed that the water capacity and mechanical properties of such hydrogels relied on the composition of the copolymer precursors. This method was also suitable for other transition metal nanoclusters, which provided a new approach to construct magnetic hydrogels.

|
Download:
|
| Fig. 12. Schematic representation of the formation of magnetic hydrogel. | |
{kind=link}
Yang et al. synthesized a novel PEO based RAFT agent to fabricate PAAPBA-b-PEO-b-PAABPA triblock copolymers based on 3-(acrylamino)phenylboronic acid (AAPBA) [134]. The gelation measurements at physiological pH demonstrated that when the PAABPA block chain length was too long, the PAAPBA-b-PEO-b-PAABPA hydrogel was unstable because of the high hydrophobicity of the PAABPA segment. Accordingly, this hydrogel dissolved within 1.5 h after the presence of high concentration of glucose, which interacted with PAABPA segment and thus increased its hydrophilicity. The in vitro test indicated that this hydrogel could achieve controlled release of FITC-BSA by the presence of glucose. Similarly, Wu et al. prepared the glucose-sensitive poly(APBA-b-LAMA) hydrogel. Initially, P(APBA) was engendered by RAFT polymerization, and subsequent as macro RAFT agent react with LAMA to gain poly(APBA-b-LAMA) hydrogel after dialyzing against water [135]. The obtained glucose-sensitive hydrogel was demonstrated good biocompatibility and applied for insulin delivery. Due to LAMA can enhance the charged phenylborates to increase the hydrophilicity of the hydrogels, the high content of hydrogels was, the high LC of insulin was. The research about insulin release showed that more LAMA contents resulted in further extending insulin release because of high cross-linking density. Without any stimulus, hydrogels behaved rapid release (~50%) 4 h before and 69% insulin released within 42h. After treated with 3mg/mL glucose, the amount of insulin release was up to 81%, which demonstrated such hydrogel was sensitive to glucose.
4. Multi-responsive hydrogels based on RAFT polymerizationBecause of the complexity of biological system, single response hydrogels often fail to meet practical application requirements, multi-responsive hydrogels have emerged on a large scale nowadays [136-138]. For multi-responsive hydrogels, it is a challenge to achieve the controlled polymerization of various functional monomers, which is the key factory for the multi-responsive performance. Obviously, RAFT polymerization provided an efficient platform to overcome such problems.
4.1. Temperature/pH dual-responsive hydrogels based on RAFT polymerizationAs temperature and pH are two important and easily controlled parameters of biological systems, temperature/pH dual-responsive hydrogels have developed into a research hotspot for biomedical applications, and the introduction here is for drug delivery systems. Most research focused temperature- and pH- responsive polymer on PNIPAAm and PAA, respectively. Compared with traditional polymerizations, RAFT polymerization provides an approach to achieve the efficiently combination of the two responsive behaviors.
Zhang et al. constructed a novel glucosyl triblock copolymer (PGNA) with narrow polydispersity based on acryloyl glucofuranose, N-isopropyl acrylamide and acrylic acid by sequential RAFT polymerization [139]. Upon temperature changes, PGNA solutions undergo a sol-gel transition, and the obtained PGNA hydrogels were also sensitive to the pH changes. The in vitro test showed that PGNA hydrogels could realize the controlled release of aspirin under the modulating of temperature and pH. This kind of temperature/pH dual-responsive hydrogels have promising potential applications as injectable drug carrier. Similarly, Yang et al. constructed ABC terpolymer (NAV) with temperature and dual-pH sensitive property based on NIAAm, AA and N, N-dimethylacrylamide (DMA) by sequential RAFT polymerization [140]. The reversible sol-gel transition could be modulated by temperature (about 37 ℃) and pH. Model drug methylene blue (MB) delineated a sustained release behavior in vitro injection of hydrogels. The tunable drug release efficiency indicated the potential of PNIPAAm- and PAA-based external sensitive hydrogels applied as drug carriers. Otherwise, as drug carriers, hydrogels were modified by chitosan based on dual responsive polymers, which got ideal release efficiency. There were many good examples. Zhang et al. employed NIAAm and AA to constructed DF-poly(NIPAAm-co-AA) as precursor by dibenzaldehyde (DF) terminated RAFT agent [141]. With the addition of glycol chitosan (GC), GC-DF-poly(NIPAAm-co-AA) hydrogel was obtained. Besides temperature and pH sensitivity, such hydrogel exhibited excellent injectable and self-healing properties. Furthermore, this kind of hydrogel has successfully applied for drug-controlled release and 3D cultivation of L929 cells. Besides, temperature- and pH-responsive hydrogel nanocomposite was prepared through RAFT polymerization of acrylic acid and N-isopropyl acrylamide onto chitosan and then modified by Hosseinzadeh et al. [142]. Such modified hydrogel nanocomposite was used as carrier for drug doxorubicin (DOX) controlled release. Notably, the in vitro release studies delineated the sustained release manner of hydrogel and the dual temperature and pH responsiveness. And within 48h, the 82% of loaded DOX was escaped from the hydrogel. From the researches mentioned above, the chitosan-based hydrogel may be a valuable drug delivery system for drug controlled released.
Wang et al. synthesized azido-PNIPAAm with pendant azido groups as hydrogel precursors by the combination of RAFT polymerization and click chemistry (Fig. 13) [143]. Then well-defined α, ω-bispropargyl PNIPAAms with various molecular weights were prepared by employing a bis propargyl terminal RAFT agent to work as cross-linker. A series of PNIPAAm based hydrogels were obtained by the click chemistry. Owing to the introduction of amine, such hydrogels could also show pH sensitivity. The swelling and stimulus response behaviors of this kind of hydrogels could be easily controlled by modulating the length of cross-linker and introducing other functional groups. Similarly, Sheng et al. [144] prepared temperature sensitive azida functionalized poly[2-(2-methoxyethoxy)ethyl methacrylate-co-oligo(ethylene glycol)methacrylate-co-N-hydroxymethyl acrylamide] and pH sensitive alkyne functionalized poly(methacrylic acid) by ATRP and RAFT polymerizations, respectively. By the click chemistry, a new temperature/pH dual-responsive hydrogel was formed. The swelling behaviors and pore size of this hydrogel could be adjusted by the temperature and pH, which directly affected the release behavior of bovine serum albumin. Besides, Chen et al. constructed a series of comb-type grafted hydrogels based on poly(N, N-dimethylaminoethyl methacrylate) (PDMAEMA) and PNIPAAm. In this study, azido end functionalized PNIPAAm–(N3)2 and PDMAEMA–(N3)2 with various chain length were prepared as macro cross-linkers by ATRP polymerization. And alkynyl side functionalized P(DMAEMA-co-ProA) and P(NIPAAm-co-ProA) were fabricated as hydrogel precursors by the RAFT polymerization of propargyl acrylate with NIPAAm or DMAEMA [145]. Through the azido-alkynyl click reaction, a temperature/pH dual-responsive hydrogel was obtained. Such hydrogels performed high swelling ration, repeatable swelling/de-swelling cycles and fast response rate under various temperature and pH conditions. The in vitro test revealed that these hydrogels could achieved a feasibility of ceftriaxone sodium depend on both temperature and pH.

|
Download:
|
| Fig. 13. Schematic illustration of the formation of PNIPAAm hydrogel. | |
{kind=link}
4.2. Other multi-responsive hydrogels based on RAFT polymerization
Except for temperature/pH dual-responsive hydrogels, RAFT polymerization was also frequently employed to construct other dual-responsive hydrogels, in which one of the stimuli was generally temperature or pH.
For example, Hemp et al. constructed a novel ABC block copolymer by sequential RAFT polymerization based on hydrophilic oligo (ethylene glycol) methyl ether methacrylate and both temperature and pH sensitive diethylene glycol methyl ether methacrylate (DEG) and [2-(methacryloyloxy) ethyl] trimethylammonium chloride [146]. The critical micelle temperatures of such copolymers were easily tuned by the composition and ionic strength of aqueous mediums. The copolymers underwent a reversible sol-gel transition upon a temperature and salt dual-depend gel point, which was helpful for biomedical applications. Liu et al. utilized RAFT polymerization to synthesize the ABC triblock copolymer that performed two-step phase transition at 29 ℃ and 39 ℃ [147]. By micelles' aggregation induced by temperature and the concentration of PEG chains, the hydrogel could be formed and show obvious thermo-responsive behavior. Banerjee et al. synthesized two kinds of block copolymers, cationic PFMA-b-PMTAC and anionic PFMA-b-PSS by sequential RAFT polymerization, respectively [148]. In water, the two kinds of micelles could be formed with PFMA as core and the rest segments as corona. The PFMA core could be crosslinked by bismaleimide via Diels–Alder (DA) click chemistry at 60 ℃ and de-crosslinked at 165 ℃. After the mix of equal charge ratio of the two oppositely charged micelles, a new class of self-assembled hydrogels could be obtained. Such hydrogel exhibited self-healing behavior upon in water (ionic interaction) or heating (DA reaction) and had potential as medical dressing. Liu and coworkers prepared the thermo- and photo-responsive hydrogels based amphiphilic triblock copolymers PNIPAAm-b-PNAM-b-PNBOC by RAFT polymerization for loading GCT and DOX [149]. The studies showed that either temperature decrease or UV irradiation at a fixed temperature lower than the critical gelation temperature could accomplish gel-to-sol transition. Drug GCT and DOX could be carried in the hydrogel, and the temperature or UV irradiation modulated the sol-gel transition to control co-release manner.
Roy et al. successfully prepared the pH and salt responsive hydrogels after Boc deprotection from a series of amino acid based organogels under acidic conditions, which were prefabricated by RAFT polymerization [150]. The swelling behavior of resulted hydrogels was greatly affected by the amino acid moiety, ionic strength and pH of solutions. Patrickios and coworkers prepared the hydrogel based on dynamic covalent polymer networks via RAFT polymerization with 2-cyano-2-propyl benzodithioate as chain transfer agent, which possessed pH and copolymer concentrations dual-responsive property [151]. The hydrogel was studied under different pH values and copolymer concentrations, and the hydrogel formation time test showed that increasing concentration could reduce the time owing to the increasing amine and acetoacetate units. Besides, when the pH value was at 8.0, such hydrogel was formed in a minimum time. Chen et al. employed RAFT polymerization to construct two kinds of zwitterionic 2-methacryloyloxyethyl phosphorylcholine-based copolymers with sugar and benzoxaborole groups, respectively [152]. By mixing the two copolymers, a hydrogel was obtained immediately based on dynamic benzoxaborole-sugar interactions, which further endowing the hydrogel self-healing and injectable abilities. The rheological tests demonstrated that the higher the sugar content and pH were, the higher strength the hydrogels possessed. Furthermore, the in vitro measurements showed that such hydrogel possessed excellent biocompatibility. This kind of self-healing hydrogels with pH/sugar dual-sensitivity have promising application potential in biomedical fields.
Recently, except for dual-response hydrogels, hydrogels with even more response properties have also been studied. For example, Voit et al. synthesized poly(4-vinylbenzoic acid) (PVBA) macromonomer by RAFT polymerization. By the radical polymerization of acrylamide functionalized PVBA and NIPAAm, novel tetra-sensitive graft copolymer hydrogels were obtained [153]. The net-PNIPAAm-g-PVBA hydrogels exhibited high volume change and sharp transition independently upon four different stimuli, temperature, pH, solvent and salt, which endowed this kind of hydrogels potential biosensor value. Additionally, Voit et al. also constructed net-PNIPAAm-g-PAA hydrogels in a similar method (Fig. 14) [154]. Such hydrogels were also sensitive to four stimuli, temperature, pH, salt and ethanol.

|
Download:
|
| Fig. 14. Schematic illustration of the formation of net-PNIPAAm-g-PAA hydrogels. | |
{kind=link}
5. Conclusion and outlook
RAFT polymerization can efficiently construct hydrogel precursors with well-defined structure, such as block copolymer, graft copolymer, star copolymer, which provides a huge library of building materials for the fabrication of intelligent hydrogels. In this review, three kinds of strategies, one-step RAFT polymerization, sequential RAFT polymerization and the combination of RAFT polymerization and click chemistry, for the preparation of intelligent hydrogels based on RAFT polymerization were summarized. Sequentially, the fabrication and biomedical applications of intelligent hydrogels with single-response and multiresponse behaviors were introduced in detail, respectively.
Although RAFT polymerization exhibited promising potential in the construction of intelligent hydrogels, some cute problems still exist. For example, RAFT agents are generally expensive and difficult to industrial production. Besides, the obtained products tend to color as the residues of RAFT agents, which is difficult to remove completely. And the residual RAFT agents may cause the products to be toxic. Furthermore, radical initiator is required in this reaction, and it is inevitable to leads to uncontrolled initiation and termination. All these problems put forward new requirements for the molecular design and preparation of RAFT agents. Although RAFT polymerization has these shortcomings, the advantages of RAFT polymerization for the preparation of intelligent hydrogels are more prominent. Maybe in the future, with the development of novel RAFT agents, the above problems can be solved to a large extent.
AcknowledgmentsThis work was supported by National Science and Technology Major Project of the Ministry of Science and Technology of China (No. 2018ZX10301402); International Cooperation and Exchange of the National Natural Science Foundation of China (No. 51820105004); the Science and Technology Program of Guangzhou (No. 201707010094); Guangdong Innovative and Entrepreneurial Research Team Program (Nos. 2013S086 and 2016ZT06S029) and the Science and Technology Planning Project of Shenzhen (No. JCYJ20170307141438157).
| [1] |
H. Yuk, B. Lu, X. Zhao, Chem. Soc. Rev. 48 (2019) 1642-1667. DOI:10.1039/C8CS00595H |
| [2] |
E.M. Ahmed, J. Adv. Res. 6 (2015) 105-121. DOI:10.1016/j.jare.2013.07.006 |
| [3] |
S.H. Zhang, P.K. Xin, Q.M. Ou, et al., J. Mater. Chem. B 6 (2018) 6723-6730. DOI:10.1039/C8TB01466C |
| [4] |
Y. Pan, Y. Gao, J.F. Shi, L. Wang, B. Xu, J. Mater. Chem. 21 (2011) 6804-6806. DOI:10.1039/c1jm10822k |
| [5] |
Z.P. Gu, H.X. Xie, C.C. Huang, L. Li, X.X. Yu, Int. J. Biol. Macromol. 58 (2013) 121-126. DOI:10.1016/j.ijbiomac.2013.03.059 |
| [6] |
J. Conde, N. Oliva, Y. Zhang, N. Artzi, Nat. Mater. 15 (2016) 1128-1138. DOI:10.1038/nmat4707 |
| [7] |
W. Lu, X.X. Le, J.W. Zhang, Y.J. Huang, T. Chen, Chem. Soc. Rev. 46 (2017) 1284-1294. DOI:10.1039/C6CS00754F |
| [8] |
Y.J. Zheng, K.Q. Huang, X.R. You, et al., Int. J. Biol. Macromol. 104 (2017) 1143-1149. DOI:10.1016/j.ijbiomac.2017.07.017 |
| [9] |
G.T. Liu, Q.J. Yuan, G. Hollett, et al., Polymer Chem. 9 (2018) 3436-3449. DOI:10.1039/C8PY00730F |
| [10] |
Z. Qian, C. Yong, L. Yu, Chin. Chem. Lett. 29 (2018) 84-86. DOI:10.1016/j.cclet.2017.07.024 |
| [11] |
S. Cai, Z. Suo, J. Mech. Phys. Solids 59 (2011) 2259-2278. DOI:10.1016/j.jmps.2011.08.008 |
| [12] |
X. Ma, Y. Li, W. Wang, Q. Ji, Y. Xia, Europ. Polym. J. 49 (2013) 389-396. DOI:10.1016/j.eurpolymj.2012.10.034 |
| [13] |
C. Yao, Z. Liu, C. Yang, et al., Adv. Funct. Mater. 25 (2015) 2980-2991. DOI:10.1002/adfm.201500420 |
| [14] |
S. Tang, R. Bhandari, S.P. Delaney, et al., Mater. Today Comm. 10 (2017) 46-53. DOI:10.1016/j.mtcomm.2016.12.003 |
| [15] |
Y. Gao, C.M. Dong, Chin. Chem. Lett. 29 (2018) 927-930. DOI:10.1016/j.cclet.2017.09.042 |
| [16] |
M. Krogsgaard, M.A. Behrens, J.S. Pedersen, H. Birkedal, Biomacromolecules 14 (2013) 297-301. DOI:10.1021/bm301844u |
| [17] |
X. Wei, Z. Hantao, Z. Chong, L. Hua, Chin. Chem. Lett. 28 (2017) 2125-2128. DOI:10.1016/j.cclet.2017.09.019 |
| [18] |
L. Liubing, G. Jun, Z. Jie, et al., ACS Appl. Mater. Interfaces 7 (2015) 8033-8040. DOI:10.1021/acsami.5b00389 |
| [19] |
J. Qu, X. Zhao, P.X. Ma, B. Guo, Acta Biomater. 58 (2017) 168-180. DOI:10.1016/j.actbio.2017.06.001 |
| [20] |
I. Lee, O. Dobre, D. Richards, et al., ACS Appl. Mater. Interfaces 10 (2018) 7765-7776. DOI:10.1021/acsami.7b18302 |
| [21] |
A.M. Rosales, K.M. Mabry, E.M. Nehls, K.S. Anseth, Biomacromolecules 16 (2015) 798-806. DOI:10.1021/bm501710e |
| [22] |
Y. Takashima, S. Hatanaka, M. Otsubo, et al., Nat. Comm. 3 (2012) 1270. DOI:10.1038/ncomms2280 |
| [23] |
D. Wu, X. Xie, A.A. Kadi, Y. Zhang, Chin. Chem. Lett. 29 (2018) 1098-1104. DOI:10.1016/j.cclet.2018.04.030 |
| [24] |
J. Thévenot, H. Oliveira, O. Sandre, S. Lecommandoux, Chem. Soc. Rev. 42 (2013) 7099-7116. DOI:10.1039/c3cs60058k |
| [25] |
E.D. Silva, P.S. Babo, R. Costa-Almeida, et al., Nanomed. -Nanotechnol. 14 (2018) 2375-2385. DOI:10.1016/j.nano.2017.06.002 |
| [26] |
F. Xiao, Y. Sun, W. Du, et al., Adv. Funct. Mater. 27 (2017) 1702147. DOI:10.1002/adfm.201702147 |
| [27] |
Y. Yang, Y. Tan, X. Wang, et al., ACS Appl. Mater. Interfaces 10 (2018) 7688-7692. DOI:10.1021/acsami.7b17907 |
| [28] |
A.V. Berezkin, F. Jung, D. Posselt, D.M. Smilgies, C.M. Papadakis, Adv. Funct. Mater. 28 (2018) 1706226. |
| [29] |
H.C. Yu, H. Zhang, K.F. Ren, et al., ACS Appl. Mater. Interfaces 10 (2018) 9002-9009. DOI:10.1021/acsami.7b18343 |
| [30] |
Z. Lei, Q. Wang, S. Sun, W. Zhu, P. Wu, Adv. Mater. 29 (2017) 1700321. DOI:10.1002/adma.201700321 |
| [31] |
L. Zhao, L. Niu, H. Liang, et al., ACS Appl. Mater. Interfaces 9 (2017) 37563-37574. DOI:10.1021/acsami.7b09395 |
| [32] |
R. Zhong, Q. Tang, S. Wang, et al., Adv. Mater. 30 (2018) 1706887. |
| [33] |
Y. Hu, C.H. Lu, W. Guo, et al., Adv. Funct. Mater. 25 (2015) 6867-6874. |
| [34] |
M. Chen, Y. Zhang, S. Jia, et al., Angew. Chem. Inter. Ed. 54 (2015) 9257-9261. DOI:10.1002/anie.201503004 |
| [35] |
J.S. Kahn, Y. Hu, I. Willner, Acc. Chem. Res. 50 (2017) 680-690. DOI:10.1021/acs.accounts.6b00542 |
| [36] |
N.K. Singh, D.S. Lee, J. Control. Release 193 (2014) 214-227. DOI:10.1016/j.jconrel.2014.04.056 |
| [37] |
R. Ghaffari, N. Eslahi, E. Tamjid, A.A. Simchi, ACS Appl. Mater. Interfaces 10 (2018) 19336-19346. DOI:10.1021/acsami.8b01154 |
| [38] |
A. Lee, Z.X. Voo, W. Chin, et al., ACS Appl. Mater. Interfaces 10 (2018) 13274-13282. DOI:10.1021/acsami.7b14319 |
| [39] |
M. Cully, Nat. Rev. Drug Discov. 14 (2015) 678-679. DOI:10.1038/nrd4744 |
| [40] |
M.M. Pakulska, K. Vulic, R.Y. Tam, M.S. Shoichet, Adv. Mater. 27 (2015) 5002-5008. DOI:10.1002/adma.201502767 |
| [41] |
T. Liu, M. Liu, S. Dou, et al., ACS Nano 12 (2018) 2818-2826. DOI:10.1021/acsnano.8b00108 |
| [42] |
M. Saito, T. Nishimura, D. Yanagida, et al., Electrochemistry 85 (2017) 637-639. DOI:10.5796/electrochemistry.85.637 |
| [43] |
D. Chatterjee, L. Llnbeck Iii, M.J. Heffernan, B.E. Munoz, A Varadhachary, US Patent App. (2019) 0111128. |
| [44] |
Z. Chen, F. Liu, Y. Chen, et al., Adv. Funct. Mater. 27 (2017) 1703036. DOI:10.1002/adfm.201703036 |
| [45] |
S.W. Seo, A.N. Enemuo, H.R. Azmand, IEEE Sensors Lett. 2 (2018) 2832006. |
| [46] |
Ž. Janićijević, F. Radovanović, Polymer 147 (2018) 56-66. DOI:10.1016/j.polymer.2018.05.065 |
| [47] |
A.E. Smith, X. Xu, C.L. McCormick, Prog. Polym. Sci. 35 (2010) 45-93. DOI:10.1016/j.progpolymsci.2009.11.005 |
| [48] |
Y. Liu, K. Xu, Q. Chang, et al., Adv. Mater. 28 (2016) 7758-7767. DOI:10.1002/adma.201601066 |
| [49] |
D.J. Keddie, Chem. Soc. Rev. 43 (2014) 496-505. DOI:10.1039/C3CS60290G |
| [50] |
Z. Huang, X. Zhang, X. Zhang, et al., Polym. Chem. 6 (2015) 607-612. DOI:10.1039/C4PY01421A |
| [51] |
G. Moad, Polym. Chem. 8 (2017) 177-219. DOI:10.1039/C6PY01849A |
| [52] |
M. Cetintas, J. De Grooth, A.H. Hofman, et al., Polym. Chem. 8 (2017) 2235-2243. DOI:10.1039/C7PY00023E |
| [53] |
M. Haqani, H. Roghani-Mamaqani, M. Salami-Kalajahi, Cellulose 24 (2017) 2241-2254. DOI:10.1007/s10570-017-1249-2 |
| [54] |
X. Fan, X. Wang, M. Cao, et al., Polym. Chem. 8 (2017) 5611-5620. DOI:10.1039/C7PY00999B |
| [55] |
J. Liu, L. Cui, N. Kong, C.J. Barrow, W. Yang, Europ. Polym. J. 50 (2014) 9-17. DOI:10.1016/j.eurpolymj.2013.10.015 |
| [56] |
Y.L. Zhang, C.K. Fu, Y.S. Li, et al., Polym. Chem. 8 (2016) 537-544. |
| [57] |
M. Szwarc, M. Levy, R. Milkovich, J. Am. Chem. Soc. 78 (1956) 192-228. |
| [58] |
J. Xu, K. Jung, A. Atme, S. Shanmugam, C. Boyer, J. Am. Chem. Soc. 136 (2014) 5508-5519. DOI:10.1021/ja501745g |
| [59] |
E.M. Ahmed, J. Adv. Res. 6 (2015) 105-121. DOI:10.1016/j.jare.2013.07.006 |
| [60] |
O.W. Webster, Science 251 (1991) 887-893. DOI:10.1126/science.251.4996.887 |
| [61] |
S. Sivaprakash, J.T. Xu, C. Boyer, Macromol. Rapid Comm. 38 (2017) 1700143. DOI:10.1002/marc.201700143 |
| [62] |
M.A. Tasdelen, M.U. Kahveci, Y. Yagci, Prog. Polym. Sci. 36 (2011) 455-567. DOI:10.1016/j.progpolymsci.2010.10.002 |
| [63] |
J.O. Zoppe, Y. Habibi, O.J. Rojas, et al., Biomacromolecules 11 (2010) 2683-2691. DOI:10.1021/bm100719d |
| [64] |
Y.G. Yu, C.C. Chae, M.J. Kim, et al., Macromolecules 51 (2018) 447-455. DOI:10.1021/acs.macromol.7b02447 |
| [65] |
Q. Yan, W. Sang, Angew. Chem. 130 (2018) 5001-5005. DOI:10.1002/ange.201712270 |
| [66] |
J.B. Zhu, E.Y.X. Chen, Angew. Chem. Int. Ed. 57 (2018) 12558-12562. DOI:10.1002/anie.201808003 |
| [67] |
X. Liu, C.G. Wang, A. Goto, Angew Chem. Int. Ed. (2019) 201814573. |
| [68] |
G. Moad, E. Rizzardo, S.H. Thang, Acc. Chem. Res. 41 (2008) 1133-1142. DOI:10.1021/ar800075n |
| [69] |
K. Matyjaszewski, J. Spanswick, Mater. Today 8 (2005) 26-33. |
| [70] |
J.D.M. Morales, R.J.S. Leija, A. Carranza, et al., Prog. Polym. Sci. 78 (2018) 139-153. DOI:10.1016/j.progpolymsci.2017.09.005 |
| [71] |
T.G. McKenzie, Q. Fu, M. Uchiyama, et al., Adv. Sci. 3 (2016) 1500394. DOI:10.1002/advs.201500394 |
| [72] |
P. Chmielarz, M. Fantin, S. Park, et al., Progr. Polym. Sci. 69 (2017) 47-78. DOI:10.1016/j.progpolymsci.2017.02.005 |
| [73] |
B.D. Fairbanks, P.A. Gunatillake, L. Meagher, Adv. Drug Deliv. Rev. 91 (2015) 141-152. DOI:10.1016/j.addr.2015.05.016 |
| [74] |
R. Nicolay, Y. Kwak, K. Matyjaszewski, Chem. Commun. (2008) 5336-5338. |
| [75] |
P. Xu, X.F. Huang, X.Q. Pan, et al., Polymers 11 (2019) 318. DOI:10.3390/polym11020318 |
| [76] |
F. Beckert, C. Friedrich, R. Thomann, R. Mulhaupt, Macromolecules 45 (2012) 7083-7090. DOI:10.1021/ma301379z |
| [77] |
J. Skey, R.K. O'Reilly, Chem. Commun. (2008) 4183-4185. |
| [78] |
Q. Fu, K. Xie, T.G. McKenzie, G.G. Qiao, Polym. Chem. 8 (2017) 1519-1526. DOI:10.1039/C6PY01994C |
| [79] |
S. Sugihara, Y. Sakamoto, M. Nakayama, K. Michishita, Y. Maeda, Polymer 154 (2018) 153-163. DOI:10.1016/j.polymer.2018.09.002 |
| [80] |
G. Moad, J. Polym. Sci. A: Polym. Chem. 57 (2019) 216-227. DOI:10.1002/pola.29199 |
| [81] |
S.J. Stace, G. Moad, C.M. Fellows, D.J. Keddie, Polym. Chem. 6 (2015) 7119-7126. DOI:10.1039/C5PY01021G |
| [82] |
B.A. Abel, C.L. Mccormick, Macromolecules 49 (2016) 465-474. DOI:10.1021/acs.macromol.5b02463 |
| [83] |
R.N. Carmean, T.E. Becker, M.B. Sims, B.S. Sumerlin, Chem 2 (2017) 13-15. DOI:10.1016/j.chempr.2016.12.010 |
| [84] |
V.H. Dao, N.R. Cameron, K. Saito, Polym. Chem. 8 (2017) 6834-6843. DOI:10.1039/C7PY01410D |
| [85] |
I. Shin, J. Nam, K. Lee, E. Kim, T.H. Kim, Polym. Chem. 9 (2018) 5190-5199. DOI:10.1039/C8PY01097H |
| [86] |
E.N. Kitiri, C.S. Patrickios, C. Voutouri, Polym. Chem. 8 (2017) 245-259. DOI:10.1039/C6PY01340F |
| [87] |
C. Chen, F.X. Kong, X.H. Wei, S.H. Thang, Chem. Commun. 53 (2017) 10776-10779. DOI:10.1039/C7CC05316A |
| [88] |
A.V. Fuchs, K.J. Thurecht, ACS Macro Lett. 6 (2017) 287-291. DOI:10.1021/acsmacrolett.7b00100 |
| [89] |
M.Y. Zhou, S.H. Liu, Y.Q. Jiang, et al., Adv. Funct. Mater. 25 (2015) 4730-4739. DOI:10.1002/adfm.201501434 |
| [90] |
Z. Li, H. Shim, M.O. Cho, et al., Carbohyd. Polym. 184 (2018) 342-353. DOI:10.1016/j.carbpol.2018.01.006 |
| [91] |
G. Fundueanu, M. Constantin, S. Bucatariu, P. Ascenzi, Polymer 110 (2017) 177-186. DOI:10.1016/j.polymer.2017.01.003 |
| [92] |
T. Wang, L.M. Chen, T.T. Shen, D.Y. Wu, Int. J. Biol. Macromol. 93 (2016) 775-782. DOI:10.1016/j.ijbiomac.2016.09.038 |
| [93] |
N. Shimada, S. Kidoaki, A. Maruyama, RSC Adv. 4 (2014) 52346-52348. DOI:10.1039/C4RA10612A |
| [94] |
X. Li, J. Zhou, Z.Q. Liu, et al., Biomaterials 35 (2014) 5679-5688. DOI:10.1016/j.biomaterials.2014.03.067 |
| [95] |
A. Navaei, D. Truong, J. Heffernan, et al., Acta Biomater. 32 (2016) 10-23. DOI:10.1016/j.actbio.2015.12.019 |
| [96] |
B.L. Ekerdt, C.M. Fuentes, Y.G. Lei, et al., Adv. Healthc. Mater. 7 (2018) 1800225. |
| [97] |
R. Elashnikov, P. Slepicka, S. Rimpelova, et al., Mater. Sci. Eng. C Mater. Biol. 9 Appl. 72 (2017) 293-300. DOI:10.1016/j.msec.2016.11.028 |
| [98] |
E. Sato Matsuo, T. Tanaka, J. Chem. Phys. 89 (1988) 1695-1703. DOI:10.1063/1.455115 |
| [99] |
L. Yong, T. Tanaka, J. Chem. Phys. 92 (1990) 1365-1371. DOI:10.1063/1.458148 |
| [100] |
X.S. Wu, A.S. Hoffman, P. Yager, J. Polym. Sci. A: Polym. Chem. 30 (1992) 2121-2129. DOI:10.1002/pola.1992.080301005 |
| [101] |
X.Z. Zhang, R.X. Zhuo, J. Colloid Interface Sci. 223 (2000) 311-313. DOI:10.1006/jcis.1999.6654 |
| [102] |
C. Löwenberg, M. Balk, C. Wischke, M. Behl, A. Lendlein, Acc. Chem. Res. 50 (2017) 723-732. DOI:10.1021/acs.accounts.6b00584 |
| [103] |
Y. Zheng, S. Zheng, React. Funct. Polym. 72 (2012) 176-184. DOI:10.1016/j.reactfunctpolym.2011.12.006 |
| [104] |
H.L. Cong, L. Li, S.X. Zheng, Polymer 54 (2013) 1370-1380. DOI:10.1016/j.polymer.2012.12.069 |
| [105] |
H.L. Cong, S.X. Zheng, Colloid Polym. Sci. 292 (2014) 2633-2645. DOI:10.1007/s00396-014-3314-9 |
| [106] |
J.G. Li, H.L. Gong, L. Li, S.X. Zheng, ACS Appl. Mater. Interfaces 6 (2014) 13677-13687. DOI:10.1021/am503148v |
| [107] |
A. Navaei, D. Truong, J. Heffernan, et al., ActaBiomater. 32 (2016) 10-23. |
| [108] |
A. Alexander, J. Khan Ajazuddin, S. Saraf, S. Saraf, Eur. J. Pharm. Biopharm. 88 (2014) 575-585. DOI:10.1016/j.ejpb.2014.07.005 |
| [109] |
Le Bohec M., M. Banère, S. Piogé, et al., Polym. Chem. 7 (2016) 6834-6842. DOI:10.1039/C6PY01639A |
| [110] |
R. Zeng, S. Xu, J. Cheng, et al., J. Appl. Polym. Sci. 131 (2013) 1082-1090. |
| [111] |
Y. Tang, S. Zhang, M. Wang, et al., J. Polym. Res. 21 (2014) 390. DOI:10.1007/s10965-014-0390-y |
| [112] |
G. Prosperiporta, B. Muirhead, H. Sheardown, J. Biomed. Mater. Res. B 105 (2017) 53-62. DOI:10.1002/jbm.b.33501 |
| [113] |
M.K. Gupta, J.R. Martin, B.R. Dollinger, M.E. Hattaway, C.L. Duvall, Adv. Funct. Mater. 27 (2017) 1704107. DOI:10.1002/adfm.201704107 |
| [114] |
H. An, K. Xu, L. Chang, et al., Polymer 147 (2018) 38-47. DOI:10.1016/j.polymer.2018.05.063 |
| [115] |
L. Mi, H. Xue, Y.T. Li, S.Y. Jiang, Adv. Funct. Mater. 21 (2011) 4028-4034. DOI:10.1002/adfm.201100871 |
| [116] |
T. Tanaka, D. Fillmore, S.T. Sun, et al., Phys. Rev. Lett. 45 (1980) 1636-1639. DOI:10.1103/PhysRevLett.45.1636 |
| [117] |
J. Qu, X. Zhao, P.X. Ma, B.L. Guo, Acta Biomater. 58 (2017) 168-180. DOI:10.1016/j.actbio.2017.06.001 |
| [118] |
S.B.D. Silva, M. Krolicka, L.A.M.V.D. Broek, A.E. Frissen, C.G. Boeriu, Carbohyd. Polym. 186 (2018) 299-309. DOI:10.1016/j.carbpol.2018.01.050 |
| [119] |
A. Shome, S. Debnath, P.K. Das, Langmuir 24 (2008) 4280-4288. DOI:10.1021/la704024p |
| [120] |
J.R. Lovett, N.J. Warren, L.P.D. Ratcliffe, M.K. Kocik, S.P. Armes, Angew. Chem. Int. Ed. 54 (2015) 1279-1283. DOI:10.1002/anie.201409799 |
| [121] |
J. Xu, D. Yang, W. Li, et al., Polymer 52 (2011) 4268-4276. DOI:10.1016/j.polymer.2011.07.015 |
| [122] |
W. Li, M. Liu, H. Chen, et al., Polym. Adv. Technol. 25 (2014) 233-239. DOI:10.1002/pat.3228 |
| [123] |
E.N. Kitiri, C.S. Patrickios, C. Voutouri, et al., Polym. Chem. 8 (2017) 245-259. DOI:10.1039/C6PY01340F |
| [124] |
Z. Guo, W. Ma, H. Gu, et al., Soft Matter. 13 (2017) 7371-7380. DOI:10.1039/C7SM00916J |
| [125] |
J. Liu, L. Cui, N. Kong, C.J. Barrow, W. Yang, Eur. Polym. J. 50 (2014) 9-17. DOI:10.1016/j.eurpolymj.2013.10.015 |
| [126] |
Y. Guo, C. Hao, X. Wang, et al., Polym. Chem. 8 (2017) 901-909. DOI:10.1039/C6PY01883A |
| [127] |
Z.T. Lu, J.Q. Zhang, Z.G. Yu, et al., New J. Chem. 41 (2017) 432-436. DOI:10.1039/C6NJ03260E |
| [128] |
D.S. Wang, M. Wagner, H.J. Butt, S. Wu, Soft Matter. 11 (2015) 7656-7662. DOI:10.1039/C5SM01888A |
| [129] |
X. Wang, C. Wang, Q. Zhang, Y. Cheng, Chem. Commun. 52 (2016) 978-981. DOI:10.1039/C5CC08391E |
| [130] |
E.D. Silva, P.S. Babo, R. Costa-Almeida, et al., Nanomed-Nanotechnol. Biol. Med. 14 (2018) 2375-2385. DOI:10.1016/j.nano.2017.06.002 |
| [131] |
Segarra-Maset M.D., V.J. Nebot, J.F. Miravet, B. Escuder, Chem. Soc. Rev. 42 (2013) 7086-7098. DOI:10.1039/C2CS35436E |
| [132] |
S.L. Banerjee, R. Hoskins, T. Swift, S. Rimmer, N.K. Singha, Polym. Chem. 9 (2018) 1190-1205. DOI:10.1039/C7PY01883E |
| [133] |
B. Jiang, W.L. Hom, X. Chen, et al., J. Am. Chem. Soc. 138 (2016) 4616-4625. DOI:10.1021/jacs.6b01271 |
| [134] |
T. Yang, X.X. Deng, F.S. Du, Z.C. Li, Acta Polym. Sin. 11 (2014) 1553-1560. |
| [135] |
C. B, L. Y, G. Q, Z. X, W. Z, Carbohyd. Res. 445 (2017) 32-39. DOI:10.1016/j.carres.2017.04.006 |
| [136] |
L. Wang, M.Z. Liu, C.M. Gao, L.W. Ma, D.P. Cui, React. Funct. Polym. 70 (2010) 159-167. DOI:10.1016/j.reactfunctpolym.2009.11.007 |
| [137] |
J.M. Knipe, N.A. Peppas, Reg. Biomater. 1 (2014) 57-65. DOI:10.1093/rb/rbu006 |
| [138] |
X. Fu, L. Hosta-Rigau, R. Chandrawati, J.W. Cui, Chem 4 (2018) 2084-2107. DOI:10.1016/j.chempr.2018.07.002 |
| [139] |
T.M. Sun, J.L. Zhu, M. Wang, et al., RSC Adv. 5 (2015) 24231-24238. DOI:10.1039/C5RA01144B |
| [140] |
C. Yan, Y. Gao, L.D. Silva, et al., Polym. Chem. 9 (2018) 4063-4072. DOI:10.1039/C8PY00838H |
| [141] |
Y.L. Zhang, C.K. Fu, Y.S. Li, et al., Polym. Chem. 8 (2017) 537-544. DOI:10.1039/C6PY01704E |
| [142] |
S. Hosseinzadeh, H. Hosseinzadeh, S. Pashaei, Z. Khodaparast, Int. J. Biol. Macromol. 121 (2019) 677-685. DOI:10.1016/j.ijbiomac.2018.10.106 |
| [143] |
J. Wang, Z. Kang, B. Qi, et al., RSC Adv. 4 (2014) 51510-51518. DOI:10.1039/C4RA07987F |
| [144] |
W.J. Sheng, T. Liu, S.X. Liu, et al., Polym. Int. 64 (2015) 1415-1424. DOI:10.1002/pi.4934 |
| [145] |
S.Q. Chen, J.M. Li, T.T. Pan, P.Y. Li, W.D. He, Polymers 8 (2016) 38. DOI:10.3390/polym8020038 |
| [146] |
S.T. Hemp, A.E. Smith, W.C. Bunyard, M.H. Rubinstein, T.E. Long, Polymer 55 (2014) 2325-2331. DOI:10.1016/j.polymer.2014.03.062 |
| [147] |
N.E. Guang, S.X. Liu, X. Li, L. Tian, H.G. Mao, Chin. J. Polym. Sci. 34 (2016) 965-980. DOI:10.1007/s10118-016-1817-1 |
| [148] |
S.L. Banerjee, N.K. Singha, Soft Matter. 13 (2017) 9024-9035. DOI:10.1039/C7SM01906H |
| [149] |
C. Wang, G. Zhang, G. Liu, J. Hu, S. Liu, J. Control. Release 259 (2017) 149-159. DOI:10.1016/j.jconrel.2016.11.007 |
| [150] |
S.G. Roy, P. De, Polymer 55 (2014) 5425-5434. DOI:10.1016/j.polymer.2014.08.072 |
| [151] |
D.E. Apostolides, A. Hadjicosta, C.S. Patrickios, Macromol. Symp. 358 (2016) 21-27. |
| [152] |
Y.J. Chen, W.D. Wang, D. Wu, et al., Biomacromolecules 19 (2018) 596-605. DOI:10.1021/acs.biomac.7b01679 |
| [153] |
D. Gräfe, S. Zschoche, D. Appelhans, B. Voit, RSC Adv. 6 (2016) 34809-34817. DOI:10.1039/C6RA01857B |
| [154] |
D. Grafe, P. Frank, T. Erdmann, et al., ACS Appl. Mater. Interfaces 9 (2017) 7565-7576. DOI:10.1021/acsami.6b14931 |