 2019, Vol. 30
2019, Vol. 30 


b State Key Laboratory of Fluid Power and Mechatronic Systems, Zhejiang University, Hangzhou 310027, China;
c MOE Key Laboratory of Macromolecular Synthesis and Functionalization, State Key Laboratory of Silicon Materials, Department of Polymer Science and Engineering, Zhejiang University, Hangzhou 310027, China
Bioactive substances are usually sensitive to external environments and encapsulation has been widely used to protect them and control their release. For example, chlorophyll, a phytolesterified magnesium porphyrin, is one of the most widely known bioactives and has a long history of uses for cancer chemoprevention, due to its anticarcinogenic and antioxidant effects [1]. However, chlorophyll alone is sensitive to light, heat, water and oxygen and easily degrades during storage, which severely limits its applications [2]. Encapsulation is thus employed to address the problem, achieving extended shelf life and controlled, targeted release of the bioactives [3-8].
Various delivery vehicles, such as nanoparticles, microcapsules, liposomes and vesicles, have been developed to encapsulate the bioactives [9-15] and have demonstrated various release modes, such as pH-triggered, light-triggered, ultrasound-triggered and electric field-triggered releases [16-22]. Despite the advancements, the delivery vehicles are mainly made of organic materials. The loose packing of the organic materials could not completely cut off the external oxygen and water vapor; the encapsulated bioactives will still slowly degrade during storage. An organicinorganic hybrid, which combines the advantages of both organic and inorganic materials, could be a promising delivery vehicle.
Single crystal with dense and highly ordered structure provides an excellent barrier to protect the encapsulated bioactives from the ambient environments. While synthetic crystals are usually homogeneous solids, but in nature, there are some inhomogeneous biogenic single crystals inside which biomacromolecules are incorporated [23]. Considerable works have been devoted to mimic the biogenic single crystals and a variety of foreign materials, such as quantum dots and polymers, are successfully incorporated inside the synthetic single crystals via gel-grown method [24-26]. The development of the crystal technique makes it possible for the functional designs of crystal composites and the exploitations of new delivery vehicles [27, 28].
Here, we develop an organic-inorganic hybrid consisted of organic nanoparticles in inorganic crystals, which improves the stability of the bioactive and implements a unique pH-triggered release. We use chlorophyll as the model bioactive. Chlorophyll is first encapsulated in shellac nanoparticles via rapid co-precipitation, which shows excellent dispersity and stability. Subsequently, we trap the nanoparticles in an agarose hydrogel and incorporate them into calcite crystals during crystallization in the gel media. The calcite crystals provide a dense shell and have greatly improved the stability of encapsulated chlorophyll. By combing the functions of organic nanoparticles and inorganic crystals, we, for the first time, achieve a unique pH-triggered release that chlorophyll could only be released by first dissolving the calcite crystal under acidic condition and then the shellac polymer under alkaline condition. The pH changes required to trigger the release exactly follow the pH changes in human gastrointestinal system, making the crystal composites an ideal delivery vehicle for targeted intestinal release. The strategy outlined in this work can be applied to design and develop different functional delivery vehicles for various applications.
To prepare chlorophyll-loaded shellac nanoparticles, chlorophyll and shellac polymer are co-dissolved in ethanol, which is quickly injected into a water reservoir using a pipet tip with a tapered nozzle, as shown in Fig. 1a. As ethanol (the good solvent) rapidly mixes with water (the poor solvent), the solubilities of both chlorophyll and shellac polymer drop dramatically. To successfully encapsulate chlorophyll in the nanoparticles, it is critical to ensure that both chlorophyll and shellac polymer experience a fast solvent exchange and co-precipitate simultaneously [29, 30]. Therefore, chlorophyll-loaded shellac nanoparticles are achieved under the rapid mixing via co-precipitation and dispersions of the nanoparticles in water are transparent and show a vivid green color, as shown in Fig. 1b.

|
Download:
|
| Fig. 1. Controlled preparation of chlorophyll-loaded shellac nanoparticles. (a) Schematic illustration of the particle preparation. Solution of chlorophyll and shellac polymer in ethanol is quickly injected into a water reservoir, which coprecipitate upon a rapid mixing of ethanol with water, forming chlorophyll-loaded nanoparticles. (b) Transparent dispersions of the nanoparticles in water, imparting a green color. (c) Linear dependence of the particle size on the concentration of shellac in ethanol. The red line is a linear fit of the experimental results. (d) A high negative zeta potential of about ζ~-40 mV observed in different sizes of nanoparticles, suggesting a good dispersion stability in water. | |
{kind=link}
The size of the nanoparticles can be tuned from 50 nm to 200 nm by varying the concentration of shellac in ethanol from 10 mg/mL to 100 mg/mL, and the size is roughly linearly proportional to the shellac concentration, as shown in Fig. 1c. The linear dependence could be attributed to the rapid mixing, under which the solvent exchange is completed before the polymers begin to aggregate. Therefore, the particle size is determined by the nucleation and growth of nanoparticles in a characteristic aggregation time and thus by the concentration of shellac polymer [29]. The nanoparticles have a relatively narrow size distribution and a relatively high negative zeta potential of ζ~-40 mV, which stabilizes their dispersion in water, as shown in Fig. 1d.
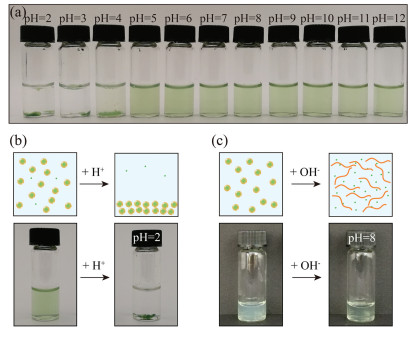
Shellac nanoparticles are promising carriers to disperse the hydrophobic chlorophyll in water over a wide pH range. Shellac polymer contains a lot of carboxylic groups and the partial dissociation of the carboxylic groups into H+ cations and RCOO- anions makes shellac nanoparticles negatively charged. The electrostatic repulsion between nanoparticles thus prevents them from aggregation. We demonstrate that the dispersions of chlorophyll-loaded shellac nanoparticles are stable in the pH range from 5 to 7, as shown in Fig. 2a. At low pH, e.g., pH 2, carboxylic groups are protonated, leading to the aggregation and sedimentation of the nanoparticles, as shown in Fig. 2b. At high pH, e.g., pH 8, more carboxylic groups are deprotonated and shellac polymer becomes soluble in water. Therefore, the nanoparticles are dissolved, releasing chlorophyll in water, which is suggested by the increase of the solution transparency in response to the pH change, as shown in Fig. 2c. Therefore, shellac nanoparticles are able to disperse chlorophyll in water at neutral or weak acidic condition and control its release under alkaline environment.

|
Download:
|
| Fig. 2. Dispersity of chlorophyll-loaded shellac nanoparticles over a wide pH range. (a) Dispersity of shellac nanoparticles at different pHs. (b) At pH 2, the carboxylic groups of shellac nanoparticles are deprotonated, leading to the aggregation and sedimentation of the nanoparticles. (c) At pH 8, shellac nanoparticles are dissolved, releasing chlorophyll in water, and the solution becomes clearer. | |
{kind=link}
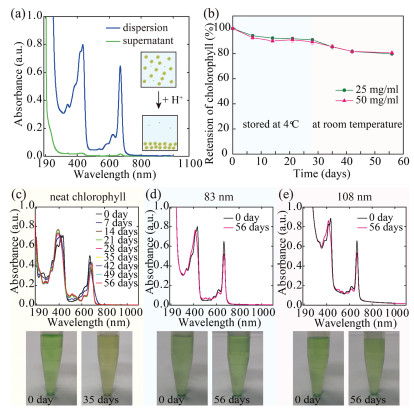
To determine the encapsulation efficiency of chlorophyll, we lower the solution pH to 2, which causes the aggregation and sedimentation of the chlorophyll-loaded nanoparticles. The amount of unencapsulated chlorophyll left in the supernatant is measured by its UV–vis absorption at 670 nm, which is linearly proportional to its concentration, as shown in Fig. 3a. We confirm that 97% chlorophyll is encapsulated in the nanoparticles and attribute the high encapsulation efficiency to the same hydrophobicity of chlorophyll and shellac. When encapsulated in the polymer matrix, shellac nanoparticles also provide a protection for chlorophyll against its degradation. To demonstrate this, we monitor the retention of chlorophyll over time by UV–vis measurements. Chlorophyll-loaded nanoparticles of d ~83 nm and d ~108 nm are prepared using 25 mg/mL and 50 mg/mL shellac in ethanol, respectively. Both samples show a retention of 80% chlorophyll after 28 days at 4 ℃ and then 28 days at room temperature, as shown in Fig. 3b. Dispersion of neat chlorophyll is prepared following the same procedure as a control experiment, whose absorption peak gradually shifts from 670 nm to 690 nm over time. Corresponding to the absorption peak shift, the color of the dispersion changes from green to yellow, which is similar to the change of green leaf to yellow leaf observed in daily life, indicating the degradation of chlorophyll, as shown in Fig. 3c. In contrast, the absorption peak of encapsulated chlorophyll at 670 nm only slightly decreases after 56 days and the dispersions show a consistent green color, as shown in Figs. 3d and e. Therefore, shellac nanoparticles could improve the stability of chlorophyll. However, the protection by shellac nanoparticles is not optimized since water and oxygen can still slowly diffuse through the polymer matrix.

|
Download:
|
| Fig. 3. High encapsulation efficiency and improved stability of chlorophyll in shellac nanoparticles. (a) UV–vis absorptions of chlorophyll loaded in shellac nanoparticles (blue curve) and unencapsulated chlorophyll left in the supernatant (green curve) when the nanoparticles aggregate and settle down at pH 2, suggesting an encapsulation efficiency of 97%. (b) Retention of chlorophyll encapsulated in shellac nanoparticles of d~83 nm (green curve) and d ~108 nm (pink curve) stored at 4 ℃ for 28 days and then at room temperature for 28 days. (c–e) UV–vis absorption spectra of unencapsulated chlorophyll and chlorophyll loaded in shellac nanoparticles of d ~83 nm and d ~108 nm, respectively. The absorption peak of unencapsulated chlorophyll shifts from 670 nm to 690 nm over time and the solution color changes from green to yellow due to the degradation of chlorophyll. In contrast, the absorption peak of encapsulated chlorophyll at 670 nm only slightly decreases over time, showing a consistent green color. | |
{kind=link}
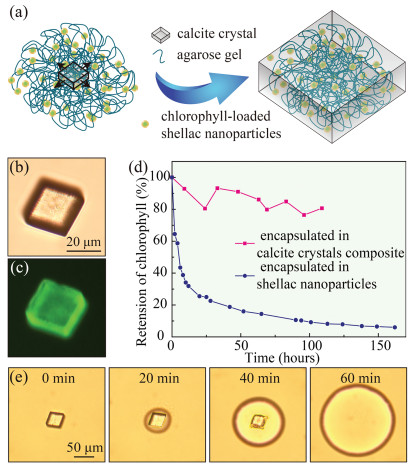
To add a second layer of protection and provide a second degree of control over the release of chlorophyll, we incorporate chlorophyll-loaded nanoparticles inside calcite crystals. Previous studies have suggested that when agarose hydrogel is mechanically strong enough, its polymer network could stand the pressure exerted by growing crystals and be incorporated inside the crystals [23]. We demonstrate that by using agarose hydrogel to fix chlorophyll-loaded nanoparticles in space, the nanoparticles could be encapsulated in calcite crystals as they grow from the agarose gel media, as schemed in Fig. 4a. Chlorophyll-loaded nanoparticles are first homogeneously mixed with the agarose solution containing 5 mmol/L CaCl2 at 40 ℃ and the mixture is left in stationary at 19 ℃ until agarose completely gels. Subsequently, 2.4 mmol/L Na2CO3 solution is added on top of the hydrogel. As a result, CO32– anions slowly diffuse into the agarose hydrogel and CaCO3 crystals form. The nanoparticles, which are trapped in the hydrogel network, are thus encapsulated in the calcite crystals, as they slowly grow in the agarose hydrogel medium. The obtained calcite crystals are directly visualized under optical microscope, as shown in Fig. 4b, and the successful incorporation of chlorophyllloaded nanoparticles in the crystal composites is confirmed by the fluorescent image shown in Fig. 4c. Calcite crystals alone do not have any fluorescence, while chlorophyll shows fluorescent property, which comes from its porphyrin ring [31]. The fluorescence of the crystal composites triggered at 488 nm and observed at 515 nm corresponds to that of chlorophyll.

|
Download:
|
| Fig. 4. Encapsulation of chlorophyll-loaded shellac nanoparticles in gel-grown calcite crystals. (a) Schematic diagram illustrating that the nanoparticles are immobilized by the agarose gel and embedded in the calcite crystal as it grows. (b) Optical and (c) fluorescent microscope images of a nanoparticle-incorporated calcite crystal. The fluorescence is attributed to the chlorophyll loaded in the nanoparticles. (d) Retention of chlorophyll under LED light when encapsulated in shellac nanoparticles (blue curve) and in calcite crystal composites (pink curve). (e) Dissolution of the calcite crystal and release of the nanoparticles under the acidic condition. | |
{kind=link}
Under the protection of calcite crystals, chlorophyll encapsulated in the crystal composites is essentially stable at room temperature in the absence of light. To demonstrate the improved stability, both chlorophyll encapsulated in the crystal composites and in the nanoparticles are tested at room temperature under a strong solar simulated irradiation. The retention of chlorophyll is monitored by measuring its fluorescent intensity, since its fluorescence disappears when it eventually degrades to nonfluorescent chlorophyll catabolites [31]. Chlorophyll is sensitive to light under aerobic conditions [32]. Less than 10% of the chlorophyll is left when encapsulated in the nanoparticles after 109 hours, as light has greatly speeded up its degradation. In contrast, 80% of the chlorophyll encapsulated in the crystal composites remains undegraded after 109 h, as shown in Fig. 4d. Therefore, the calcite crystals with dense and highly ordered structure provide an excellent barrier to protect chlorophyll from harsh ambient environments, resulting in greatly enhanced stability.
The presence of calcite crystals provides yet another degree of control over the release of chlorophyll, which is particular important to prevent the leakage and design the release. When chlorophyll is encapsulated in shellac nanoparticles alone, it will be released if the pH was accidentally raised up to an alkaline condition during storage. However, chlorophyll encapsulated in the crystal composites could only be released following the exact pH changes, i.e., the pH first changes to acidic, under which the calcite crystals are dissolved, and then to alkaline, under which shellac nanoparticles become soluble. To demonstrate the unique pH-triggered release mode of the crystal composites, we use HCl as the stimulus and trigger the release of chlorophyll-loaded nanoparticles from the calcite crystals, as shown in Fig. 4e, and use NaOH as the stimulus and trigger the release of chlorophyll from the nanoparticles, as shown in Fig. 2c. The pH changes required to trigger the release is similar to human gastrointestinal system that the pH first changes to acidic in the stomach (pH ~3.5) and then to alkaline in the intestine (pH ~7.5), making the crystal composites an ideal delivery vehicle for controlled, targeted intestinal release.
We develop a novel delivery vehicle consisted of organic nanoparticles in inorganic crystals and successfully encapsulated chlorophyll in the crystal composites, which demonstrates excellent stability and controlled, targeted intestinal release. Chlorophyll-loaded nanoparticles are first prepared by co-precipitation and then incorporated in calcite crystals by gel-grown method. Both shellac polymer and calcite crystal are FDAapproved, making the crystal composites suitable for food and drug delivery. Under the protection of shellac nanoparticles and calcite crystals, chlorophyll shows an excellent stability performance and could only be released by first dissolving calcite crystals under acidic condition and then dissolving shellac nanoparticles under alkaline condition. The unique pH-triggered release, which requires a pH change similar to that of human gastrointestinal system, is well suited for controlled, targeted intestinal release. The crystal composites of organic nanoparticles in inorganic crystals developed in this work also offers a unique platform to design functional delivery vehicles for various applications.
AcknowledgmentsThis work is supported by the National Natural Science Foundation of China (Nos. 21878258, 11704331 and 51625304), "the Fundamental Research Funds for the Central Universities" (No. 2018QNA4046).
Appendix A. Supplementary dataSupplementary material related to this article can befound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.08.007.
| [1] |
M.Q. Chu, H.K. Li, Q. Wu, et al., Biomaterials 35 (2014) 8357-8373. DOI:10.1016/j.biomaterials.2014.05.049 |
| [2] |
S.J. Schwartz, T.V. Lorenzo, J. Food Sci. 56 (1991) 1059-1062. DOI:10.1111/j.1365-2621.1991.tb14641.x |
| [3] |
X.L. Yang, X.J. Ju, X.T. Mu, et al., ACS Appl. Mater. Interfaces 8 (2016) 10524-10534. DOI:10.1021/acsami.6b01277 |
| [4] |
Y.H. Geng, X.H. Ge, S.B. Zhang, et al., Lab Chip 18 (2018) 1838-1843. DOI:10.1039/C8LC00293B |
| [5] |
B. Kim, S. Lee, S.H. Kim, Adv. Mater. Interfaces 5 (2018) 1701472. DOI:10.1002/admi.201701472 |
| [6] |
S. Zhou, F. Jing, S.S. Datta, et al., Adv. Funct. Mater. 23 (2013) 5925-5929. DOI:10.1002/adfm.201301030 |
| [7] |
C.Y. Hu, Z. Chen, S.J. Wu, et al., Chin. Chem. Lett. 28 (2017) 1905-1909. DOI:10.1016/j.cclet.2017.07.020 |
| [8] |
L. Kong, R. Chen, X. Wang, et al., Lab Chip 19 (2019) 2089-2095. DOI:10.1039/C9LC00240E |
| [9] |
L.Y. Chu, T. Yamaguchi, S. Nakao, Adv. Mater. 14 (2002) 386-389. DOI:10.1002/1521-4095(20020304)14:5<386::AID-ADMA386>3.0.CO;2-I |
| [10] |
L.L. Kong, E. Amstad, M.T. Hai, et al., Chin. Chem. Lett. 28 (2017) 1897-1900. DOI:10.1016/j.cclet.2017.07.017 |
| [11] |
Y.C. Su, H. Zhao, J.R. Wu, J.H. Xu, RSC Adv. 6 (2016) 112292-112299. DOI:10.1039/C6RA19048K |
| [12] |
A. Vian, E. Amstad, Soft Matter 15 (2019) 1290-1296. DOI:10.1039/C8SM02047G |
| [13] |
Z. Fang, L.Y. Wan, L.Y. Chu, et al., Expert Opin. Drug Deliv. 12 (2015) 1943-1953. DOI:10.1517/17425247.2015.1071352 |
| [14] |
X.R. You, X.J. Ju, F. He, et al., ACS Appl. Mater. Interfaces 9 (2017) 19258-19268. DOI:10.1021/acsami.7b05701 |
| [15] |
C.Z. Sun, Y. Liang, M.Y. Zhao, et al., J. Biomed. Nanotechnol. 13 (2017) 946-959. DOI:10.1166/jbn.2017.2411 |
| [16] |
J. Wei, X.J. Ju, X.Y. Zou, et al., Adv. Funct. Mater. 24 (2014) 3312-3323. DOI:10.1002/adfm.201303844 |
| [17] |
S. Lee, T.Y. Lee, E. Amstad, S.H. Kim, Adv. Mater. Technol 3 (2018) 1800006. DOI:10.1002/admt.201800006 |
| [18] |
E. Amstad, S.H. Kim, D.A. Weitz, Angew. Chem. Int. Ed. 51 (2012) 12499-12503. DOI:10.1002/anie.201206531 |
| [19] |
E. Amstad, J. Kohlbrecher, E. Muller, et al., Nano Lett. 11 (2011) 1664-1670. DOI:10.1021/nl2001499 |
| [20] |
S. Kennedy, J. Hu, C. Kearney, et al., Biomaterials 75 (2016) 91. DOI:10.1016/j.biomaterials.2015.10.008 |
| [21] |
R. Hu, H. Zheng, J. Cao, et al., J. Biomed. Nanotechnol. 13 (2017) 1058-1068. DOI:10.1166/jbn.2017.2406 |
| [22] |
Z. Sun, C. Yang, M. Eggersdorfer, et al., Chin. Chem. Lett. (2019). DOI:10.1016/j.cclet.2019.04.040 |
| [23] |
E. Asenath-Smith, H.Y. Li, E.C. Keene, et al., Adv. Funct. Mater. 22 (2012) 2891-2914. DOI:10.1002/adfm.201200300 |
| [24] |
Y.J. Liu, W.T. Yuan, Y. Shi, et al., Angew. Chem. Int. Ed. 53 (2014) 4127-4131. DOI:10.1002/anie.201310712 |
| [25] |
Y.J. Liu, H.D. Zang, L. Wang, et al., Chem. Mater. 28 (2016) 7537-7543. DOI:10.1021/acs.chemmater.6b03589 |
| [26] |
Y.J. Liu, K. He, W.T. Yuan, et al., Chin. Chem. Lett. 29 (2018) 1666-1670. DOI:10.1016/j.cclet.2018.05.044 |
| [27] |
Y.J. Liu, Y. Tan, J. Ren, et al., Chin. Chem. Lett. 29 (2018) 1296-1300. DOI:10.1016/j.cclet.2018.07.005 |
| [28] |
H.B. Li, G.B. Xue, J.K. Wu, et al., Chin. Chem. Lett. 28 (2017) 2121-2124. DOI:10.1016/j.cclet.2017.08.011 |
| [29] |
J.M. Lim, A. Swami, L.M. Gilson, et al., ACS Nano 8 (2014) 6056-6065. DOI:10.1021/nn501371n |
| [30] |
L.D. Scampavia, G. Blankenstein, J. Ruzicka, G.D. Christian, Anal. Chem. 67 (1995) 2743-2749. DOI:10.1021/ac00113a004 |
| [31] |
S. Hortensteiner, Annu. Rev. Plant Biol. 57 (2006) 55-77. DOI:10.1146/annurev.arplant.57.032905.105212 |
| [32] |
E. Lee, H. Ahn, E. Choe, Food Sci. Biotechnol. 23 (2014) 1061-1065. DOI:10.1007/s10068-014-0145-x |