 2019, Vol. 30
2019, Vol. 30 


b State Key Laboratory of Advanced Technology for Materials Synthesis and Processing, Wuhan University of Technology, Wuhan 430070, China
The rapid developments of renewable energy harvesting and electric vehicles lead to the growing demand for advanced energy storage devices with high energy densities. In the past decades, as a promising energy storage device, lithium oxygen batteries (Li—O2 batteries) have attracted extensive interests worldwide due to their ultrahigh theoretical energy density of around 3500 Wh/kg based on the reversible formation and decomposition of Li2O2 [1–7]. However, the practical application of Li—O2 batteries is still hindered by several critical challenges including high overpotential, low round-trip efficiency, poor rate capability, and short cycle life [8–11]. These challenges mainly result from the sluggish oxygen reduction reaction (ORR) and oxygen evolution reaction (OER) in the oxygen diffusion electrodes, as well as from the deposition and incomplete decomposition of insulating and insoluble Li2O2. Therefore, the rationally designed porous archi-tectures of oxygen diffusion electrodes are highly desired to offer abundant catalytically active sites for both ORR and OER, sufficient inner space for accommodating the discharge product Li2O2, and developed channels for maintaining the efficient transport of O2, Li+, and electrons [12, 13].
In practice, the porous architectures of oxygen electrodes are generally realized by compressing the composites of porous carbon materials and binders [14]. Porous carbon is a preferred cathode material because of its high electronic conductivity, potential catalytic activity for ORR and OER, adjustable pore structure and surface area, and low cost [15–18]. In previous studies, in order to optimize the performance of carbonaceous cathode, the effects of porosities and surface areas on the performance of porous carbon were comprehensively investigated. Some researchers suggested that the discharge capacities of Li—O2 batteries are proportional to the externally accessible surface areas of carbonaceous cathodes [19, 20]. Whereas, the others demonstrated that the initial discharge capacities are directly correlated with the pore volume and the distribution of pore sizes instead of the surface areas [21–23]. Besides, the surface chemical composition of carbonaceous cathodes could also be a significant but usually ignored factor that strongly influences the performance of Li—O2 batteries. So far, only Xiao et al. had demonstrated that the defects and functional groups on graphene favor the formation of isolated nanosized Li2O2 particles and help prevent air blocking in the air electrode via scanning electronic microscopy observations coupled with theoretical calculation [24]. Generally, it is very difficult to directly observe the surface chemistry effects of carbonaceous oxygen electrodes on the Li—O2 batteries capacities, since the intrinsic physicochemical properties of carbon materials are probably shielded by binders and other additives in a composite electrode. Thus, the surface chemistry effects on the discharge capacities of carbonaceous cathodes are still unclear and should be further studied with novel research strategies.
In this work, therefore, we propose chemically activated carbon cloth (CACC) as an ideal model to study the effect of surface functional groups on the discharge capacities of carbonaceous oxygen electrodes for Li—O2 batteries. A piece of carbon cloth (CC) can function as a gas diffusion electrode without binders because of its porous architecture weaved of crisscrossed carbon fibers. All the surfaces of the carbon fibers are easily accessible to the reaction molecules. More importantly, the intrinsic surface chemistry effects on the performance of carbonaceous cathode could be directly observed without the influences of binders and additives. The results in the present work indicate that the introduction of surface carboxyl group (—COOH) significantly enhances the discharge capacities of carbon cloth electrodes. This direct observation provides a new opportunity to further optimize the performance of carbonaceous oxygen electrodes in Li–O2 batteries via appropriate surface functional group engineering.
The CACC was prepared by a hydrothermal treatment followed by annealing in Ar/H2 atmosphere. Firstly, a piece of commercially available CC (2.0×2.0 cm2, Cetech Crop.) was sequentially cleaned by acetone, alcohol, and deionized water. Then, the cleaned CC was treated in a 100 mL Teflon-lined autoclave at 200 ℃ for 24 h with a mixture solution of 50 mL isopropanol, 5 mL hydrofluoric acid solution (40 wt%) and 5 mL distilled water. After that, the obtained CACC was washed with deionized water three times, and then annealed at 600 ℃ for 3 h in an Ar/H2 (95%/5%) atmosphere.
Scanning electron microscopy (SEM, Zeiss Ultra Plus) was used to investigate the morphologies of the as-prepared cathodes before and after discharge. The X-ray diffraction (XRD) patterns of CC and CACC were recorded on a Rigaku D/MAX-RBU-200B X-ray diffractometer using Cu Kα (λ = 0.154 nm) radiation with a scan rate of 5 ℃/min. The surface chemical compositions of the prepared samples before and after discharge were investigated by X-ray photoelectron spectroscopy (XPS, VG Multilab2000X), and Raman Spectroscopy (Raman, Invia). A dye-absorption method [25, 26] was adopted to compare the accessible surface areas of CC and CACC. In detail, the Ultraviolet-visible spectroscopies (UV–vis, N4) of methylene blue (MB) aqueous solutions before and after the absorptions by CC and CACC were collected, respectively. Further-more, the electrochemically active surface areas (ECSA) of CC and CACC were determined by the double-layer capacitive during the repeated cyclic voltammetry (CV) scanning.
The Li—O2 batteries were assembled into R2032 coin cells with a lithium foil (with the diameter of 1.56 cm and the thickness of 1.5 mm) as the anode, a glass fiber separator (GF/D, Whatman), and 1 mol/L lithium bis(trifluoromethane) sulfonamide (LiTFSI) in tetraethylene glycol dimethyl ether (TEGDME) as the electrolyte. The pristine CC and CACC were cut into 1.0×1.0 cm2 squares and used as the cathodes, respectively. Holes (diameter of 1 mm, 21 holes) were punched in the bottom canister of the coin cells for oxygen flow. All the operations were conducted in an Argon-filled glove box with O2 and H2O levels less than 0.1 ppm. The prepared cells were put into an oxygen container with the pressure of 1 atm. Galvanostatic discharge tests for the as-prepared Li–O2 batteries were conducted on a Land CT3008W battery testing system at room temperature. The discharged cathodes were disassembled from the coin cells, and then washed with TEGDME several times. Before further investigations, the discharged cathodes were dried and kept in the glove box.
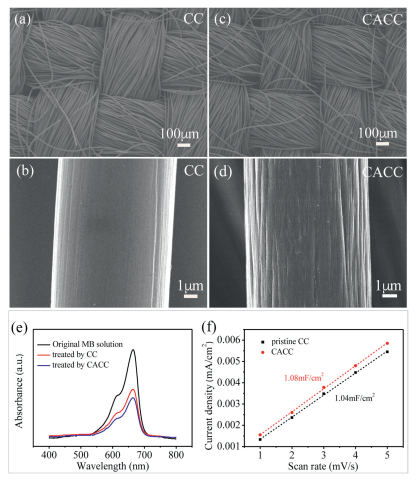
The morphologies and surface areas of the carbon clothes before and after the chemical treatment were firstly investigated. Figs. 1a-d compare the SEM images of CC and CACC. As shown in Fig. 1a, the commercially available carbon cloth is weaved of interlaced carbon fibers. Under a higher magnification in Fig. 1b, the carbon fibers in CC exhibit a relatively smooth surface. After the chemical treatment, the interweaving architecture of CACC in Fig. 1c remains to function as a porous electrode. However, the carbon fibers (Fig. 1d) in CACC become a little rougher than those in pristine CC, indicating that the carbon fibers are corroded to some extent. Furthermore, a dye-absorption method was used to determine the surface area changes of carbon clothes before and after the chemical treatment. Fig. 1e presents the UV–vis absorption spectra from the MB solutions before and after the adsorptions of the pristine CC and CACC, respectively. It can be found that the absorption intensity of the MB solution treated by CACC is slightly lower than that by CC. This means that more MB dye molecules are absorbed by CACC than by pristine CC. Therefore, the chemical treatment slightly enhances the accessible surface areas of carbon clothes (from CC to CACC) via a currently unknown mechanism. The slightly enhanced surface area of CACC is also confirmed by the determination of electrochemically active surface area. The non-faradic capacitive current responses of CC and CACC in the cyclic voltammetry tests at a double-layer region are recorded at different scan rates in Fig. S1 (see the Supporting infroamtion for the figure and experimental details). Fig. 1f presents the plots of apparent current densities (calculated by dividing the geometric area) versus the scan rates of both CC and CACC. The slopes of these linear plots provide the double-layer capacitances of the electrodes. As presented in Fig. 1f, the capacitances of CC and CACC are determined to be 1.04 and 1.08 m F/cm2, respectively. By assuming that the capacitance of an ideal plate electrode is 60 mF/μm2 [27–29], the roughness factors of CC and CACC are calculated to be 17.33 and 18.00, respectively. In summary, all the above analyses indicate that the carbon fibers in CC are corroded by the chemical treatment, and the specific surface area of CACC is not significantly enhanced compared with pristine CC.

|
Download:
|
| Fig. 1. The SEM images of (a, b) CC and (c, d) CACC. (e) UV–vis absorption spectra collected for the original MB solution and for the MB solutions after the adsorptions of CC and CACC. (f) The plots of current densities versus scan rates in the CV tests for CC and CACC. | |
{kind=link}
Besides the morphologies and surface areas, the surface chemical compositions of CC and CACC were also comparatively studied by XPS, Raman spectroscopy, and XRD. Figs. 2a and b compare the high resolution XPS spectra of CC and CACC in the C 1s region. The fitted major C 1s peaks at 284.6 eV for both CC and CACC are attributed to the adventitious hydrocarbon, which is usually used as a binding energy reference. The peaks at 285.6 eV for CC and CACC are ascribed to the appearance of C—C bands. Compared with CC, CACC has an additional peak at 288.7 eV for carboxyl carbon [25, 30]. This result indicates that CACC is oxidized to some extent. In Fig. 2c, the O 1s spectrum of the pristine CC only shows a single peak at 532.4 eV corresponding to the —OH group absorbed on the carbon surface. By contrast, the broader O 1s spectrum of CACC could be fitted as three distinct peaks that are absorbed —OH at 532.4 eV, C=O at 531.6 eV, and —OH in —COOH at 534.2 eV (Fig. 2d) [31]. Therefore, the chemical treatment results in the oxidation of the carbon surface to produce —COOH groups. The Raman spectroscopy studies further confirm this observation. The Raman spectra of both CC and CACC in Fig. 2e clearly show the distinct D and G bands of carbon materials. It is found that the intensity ratio of D and G peaks (ID/IG) of CACC (1.17) is higher than that of pristine CC (1.04). The enhanced ID/IG of CACC indicates a higher degree of disorder coupled with more functional groups on its surface. Finally, XRD patterns were collected for CC and CACC to further confirm their chemical components. As shown in the Fig. 2f, all the diffraction peaks of CC and CACC could be indexed to graphite phase, and the peaks at around 25° and 45° are assigned to (002) and (101) planes, respectively. In conclusion, although the chemical treatment does not significantly change the surface area of carbon cloth, it introduces —COOH groups on the carbon surface with graphite phase.

|
Download:
|
| Fig. 2. C 1s core-level XPS spectra of (a) CC and (b) CACC. O 1s core-level XPS spectra of (c) CC and (d) CACC. (e) Raman spectra of CC and CACC. (f) XRD patterns of CC and CACC. | |
{kind=link}
As discussed above, CC and CACC have very similar surface areas. Therefore, according to previous reports, CC and CACC might be expected to exhibit similar discharge capacities. In the present work, the Li—O2 cells utilizing pristine CC and CACC as the oxygen electrodes respectively are discharged at the current density of 0.05 mA/cm2 with a cut-off voltage of 2.2 V. Surprisingly, as shown in Figs. 3a and b, CACC shows a significantly enhanced discharge capacity of 1.23 mAh/cm2 after the chemical treatment, which is nearly 2.6 times as much as the capacity of the pristine CC (~0.48 mAh/cm2). This result indicates that the surface functional group should be a critical factor that influences the discharge capacity of carbonaceous oxygen electrodes beside the surface area, since the main difference of CC and CACC is the surface functional group. Herein, we make the assumption that the introduction of —COOH group on the surfaces of carbonaceous materials facilitates the growth of Li2O2 and consequently enhances the discharge capacity of CACC.

|
Download:
|
| Fig. 3. The voltage profiles of (a) CC and (b) CACC; SEM images of fully discharged CC (c, d) and CACC (e, f) (at a cut-off potential of 2.2 V, 0.05 mA/cm2). | |
{kind=link}
In order to confirm the assumption of the effect of —COOH group, the discharged cathodes were disassembled from the cells discharged to a cut off potential of 2.2 V and then investigated by XPS and SEM. XPS studies in Fig. S2 (Supporting information) indicate that the main products on both the discharged CC and CACC are Li2O2 [32, 33]. Figs. 3c–f compare the SEM images of discharged electrodes. In Figs. 3c and d, loose and irregular particles are observed on the discharged CC, which results in the relatively lower discharge capacity of CC. By contrast, discharged CACC in Figs. 3e and f show a closely packing of toroid-shaped particles. Moreover, there are also additional needle-like discharge products on the edges of CACC fibers. Therefore, the surface carboxyl groups on CACC might enable the packing of toroid-shaped Li2O2 to achieve the enhanced capacity.
Regarding the growth of Li2O2 in the oxygen diffusion electrodes, two competitive mechanisms were proposed in previous reports. Some previous works suggested that oxygen is first reduced to LiO2 on the electrode surface, and then the adsorbed LiO2 will be further reduced to product Li2O2 through a surface mechanism. If so, the fibers in the carbon clothes will be conformally coated by the discharge products. The surface mechanism is not consistent with the results in the present work. By contrast, in a solution mechanism, oxygen should be reduced to the solvated LiO2, and then LiO2 dissolved in the solvent disproportionate to Li2O2 and O2. The SEM images of discharged CC and CACC support the solution mechanism, in which the nucleation sites for Li2O2 is critical for the discharge capacity. On the CC cathode, only scared nucleation sites are provided for the formation of Li2O2. Therefore, low capacity of CC is observed. However, on the CACC cathode, the chemical treatment has introduced abundant —COOH groups onto the electrode surface, which function as appropriate nucleation sites for the nucleation-growth process of Li2O2.
To further confirm the mechanism, CC and CACC were discharged at 0.05 mA/cm2 to a cut-off capacity of 0.1 mAh/cm2 were investigated by SEM. As shown in Figs. 4a and b, only few Li2O2 particles are found on the CC surface. In contrast, Figs. 4c and d clearly show that Li2O2 particles are uniformly distributed on the surface of CACC. These initially formed Li2O2 particles act as the nucleation sites for the further discharge, and lead to a significantly enhanced capacity of CACC. In addition to the solution mechanism and nucleation-growth process, the particle sizes of Li2O2 are also regulated by the surface functional groups. When the small Li2O2 aggregation is formed, the electrode surface will be passivated early and thus yield a low capacity. On the contrary, the formation of larger Li2O2 aggregation will prolong the passivation of electrode surface and lead to a relatively high capacity [34–36]. In the present work, as shown in Figs. 3e and f, the formation of toroid-like Li2O2 on CACC surface makes the passivation of CACC surface slower, thus achieving a higher capacity than the pristine CC.

|
Download:
|
| Fig. 4. SEM images of discharged (with a specific charge capacity of 0.1 mA h/cm2) CC (a, b) and CACC (c, d). | |
{kind=link}
In summary, utilizing carbon cloth as the ideal model, we have directly observed that the introduction of surface carboxyl groups enhances the capacities of carbonaceous oxygen diffusion electro-des for aprotic lithium–oxygen batteries. The carboxyl groups not only function as the appropriate nucleation sites for Li2O2 but also induce the formation of toroid-like Li2O2. The closely packing of toroid-shaped particles on chemically activated carbon clothes provide enhanced capacities. This work provides a promising way to further optimize the carbonaceous oxygen diffusion electrodes via surface functional group engineering.
AcknowledgmentsThis work was supported by grants from the National Natural Science Foundation of China (Nos. 21673169, 51672205), the National Key R & D Program of China (No. 2016YFA0202602), the Research Start-Up Fund from Wuhan University of Technology, and the Fundamental Research Funds for the Central Universities (WUT: Nos. 2019IB003, 2016IVA083).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.07.011.
| [1] |
G. Girishkumar, B. McCloskey, A.C. Luntz, S. Swanson, W. Wilcke, J. Phys. Chem. Lett. 1 (2010) 2193-2203. DOI:10.1021/jz1005384 |
| [2] |
P.G. Bruce, S.A. Freunberger, L.J. Hardwick, J.M. Tarascon, Nat. Mater. 11 (2012) 19-29. DOI:10.1038/nmat3191 |
| [3] |
C. Wu, C.B. Liao, L. Li, J. Yang, Chin. Chem. Lett. 27 (2016) 1485-1489. DOI:10.1016/j.cclet.2016.03.023 |
| [4] |
P. Tan, H.R. Jiang, X.B. Zhu, et al., Appl. Energy 204 (2017) 780-806. DOI:10.1016/j.apenergy.2017.07.054 |
| [5] |
W. Chen, Y.F. Gong, J.H. Liu, Chin. Chem. Lett. 28 (2017) 709-718. DOI:10.1016/j.cclet.2016.10.023 |
| [6] |
B. Scrosati, Y.K. Sun, Y.K. Sun, Energy Environ. Sci. 4 (2011) 3287-3295. DOI:10.1039/c1ee01388b |
| [7] |
Z. Chen, Q. Wang, X. Zhang, et al., Sci. Bull. (Beijing) 63 (2018) 548-555. DOI:10.1016/j.scib.2018.04.003 |
| [8] |
L. Zhao, Q. Wang, X. Zhang, et al., ACS Appl. Mater. Inter. 10 (2018) 35888-35895. DOI:10.1021/acsami.8b09197 |
| [9] |
T. Li, C. Wu, H. Yuan, L. Li, J. Yang, Chin. Chem. Lett. 28 (2017) 2155-2158. DOI:10.1016/j.cclet.2017.09.021 |
| [10] |
F.J. Li, T. Zhang, H.S. Zhou, Energy Environ. Sci. 6 (2013) 1125-1141. DOI:10.1039/c3ee00053b |
| [11] |
J.L. Shui, J.S. Okasinski, P. Kenesei, et al., Nat. Commun. 4 (2013) 2255-2261. DOI:10.1038/ncomms3255 |
| [12] |
C. Tran, X.Q. Yang, D.Y. Qu, J. Power Sources 195 (2010) 2057-2063. DOI:10.1016/j.jpowsour.2009.10.012 |
| [13] |
P. Zhang, Y. Zhao, X.B. Zhang, Chem. Soc. Rev. 47 (2018) 2921-3004. DOI:10.1039/C8CS00009C |
| [14] |
J.G. Zhang, D. Wang, W. Xu, J. Xiao, R.E. Williford, J. Power Sources 195 (2010) 4332-4337. DOI:10.1016/j.jpowsour.2010.01.022 |
| [15] |
P. Kichambare, J. Kumar, S. Rodrigues, B. Kumar, J. Power Sources 196 (2011) 3310-3316. DOI:10.1016/j.jpowsour.2010.11.112 |
| [16] |
J. Li, H. Zhang, Y. Zhang, et al., Nanoscale 5 (2013) 4647-4651. DOI:10.1039/c3nr00337j |
| [17] |
S. Ryu, B.G. Kim, J.W. Choi, H. Lee, J. Nanosci. Nano Technol. 15 (2015) 7611-7614. DOI:10.1166/jnn.2015.11160 |
| [18] |
X.B. Zhu, Y. Yan, W. Wan, et al., Mater. Lett. 215 (2018) 71-74. DOI:10.1016/j.matlet.2017.12.056 |
| [19] |
S. Meini, M. Piana, H. Beyer, J. Schwämmlein, H.A. Gasteiger, J. Electrochem. Soc. 159 (2012) A2135-A2142. DOI:10.1149/2.011301jes |
| [20] |
D.Y. Zhai, H.H. Wang, J.B. Yang, et al., J. Am. Chem. Soc. 135 (2013) 15364-15372. DOI:10.1021/ja403199d |
| [21] |
Y. Li, J. Wang, X. Li, et al., Chem. Commun. (Camb.) 47 (2011) 9438-9440. DOI:10.1039/c1cc13464g |
| [22] |
J. Tang, J. Liu, C. Li, et al., Angew. Chem. Int. Ed. 54 (2015) 588-593. |
| [23] |
S.B. Ma, D.J. Lee, V. Roev, D. Im, S.G. Doo, J. Power Sources 244 (2013) 494-498. DOI:10.1016/j.jpowsour.2013.03.150 |
| [24] |
J. Xiao, D. Mei, X. Li, et al., Nano Lett. 11 (2011) 5071-5078. DOI:10.1021/nl203332e |
| [25] |
W. Wang, W. Liu, Y Zeng, et al., Adv. Mater. 27 (2015) 3572-3578. DOI:10.1002/adma.201500707 |
| [26] |
D. Ye, Y. Yu, J. Tang, L. Liu, Y. Wu, Nanoscale 8 (2016) 10406-10414. DOI:10.1039/C6NR00606J |
| [27] |
W. Xu, Z. Lu, X. Lei, Y. Li, X. Sun, Phys. Chem. Chem. Phys. 16 (2014) 20402-20405. DOI:10.1039/C4CP02952F |
| [28] |
Y. Yu, J. Zhang, M. Zhong, S. Guo, Electrocatalysis 9 (2018) 653-661. DOI:10.1007/s12678-018-0473-3 |
| [29] |
B. Lu, D. Cao, P. Wang, G. Wang, Y. Gao, Int. J. Hydrogen. Energ. 36 (2011) 72-78. DOI:10.1016/j.ijhydene.2010.09.056 |
| [30] |
L. Xiao, E. Li, J. Yi, et al., J. Alloys. Compd. 764 (2018) 545-554. DOI:10.1016/j.jallcom.2018.06.081 |
| [31] |
W. Li, T. Wu, S. Zhang, et al., Chem. Commun. (Camb.) 54 (2018) 11188-11191. DOI:10.1039/C8CC06000B |
| [32] |
J. Luo, X. Yao, L. Yang, et al., Nano Res. 10 (2017) 4318-4326. DOI:10.1007/s12274-017-1660-x |
| [33] |
J. Xie, X. Yao, Q. Cheng, et al., Angew. Chem. Int. Ed. 54 (2015) 4299-4303. DOI:10.1002/anie.201410786 |
| [34] |
B.D. Adams, C. Radtke, R. Black, et al., Energy Environ. Sci. 6 (2013) 1772-1778. DOI:10.1039/c3ee40697k |
| [35] |
C. Shen, Z. Wen, F. Wang, et al., J. Power Sources 294 (2015) 593-601. DOI:10.1016/j.jpowsour.2015.06.130 |
| [36] |
B. Horstmann, B. Gallant, R. Mitchell, et al., J. Phys. Chem. Lett. 4 (2013) 4217-4222. DOI:10.1021/jz401973c |