 2019, Vol. 30
2019, Vol. 30 


b College of Chemistry, Henan Key Laboratory of Chemical Biology and Organic Chemistry, Key Laboratory of Applied Chemistry of Henan Universities, Zhengzhou University, Zhengzhou, China
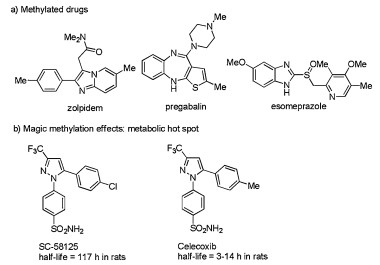
Methylation involved in many fundamental biological process-es, such as DNA replication, transcription, and epigenetics [1]. Also, the methyl group is one of the most prevalent functionalities in biologically active molecules (Fig. 1a). This simplest alkyl fragment can modulate both the biological and physical properties of a molecule, such as solubility, selectivity and metabolic half-life [2]. As was the case with anti-inflammatory drug celecoxib, a methyl group was introduced to decrease the half-life, (Fig. 1b) [3]. In light of these merits, catalytic methods to attach methyl group are particularly important to develop.

|
Download:
|
| Fig. 1. Examples of methylated drugs and a magic methyl effect. | |
The most extensive studies on the direct methylation of C–H bond involve the use of MeI or organometallic reagents [4], such as Me4Sn, MeB(OH)2, MeMgCl, Me3Al, and Me2Zn. Since the seminal work of Minisci in 1971 on the addition of the nucleophilic methyl radical to electron-deficient heterocycles, substantial progress has been made in radical C–H methylation [5]. Some methyl radical precursors are known, such as HOAc [5], peroxide [6], DMSO [7], methanol [8], and others [9]. Unfortunately, these one-step methylation methodologies are limited in their application owing to the use of strong stoichiometric oxidants or high temperature to generate the requisite methyl radical. A great difficulty arises from the similar polarities of the starting material and the methylated product, which makes purification difficult. Recently, a two-step radical methylation becomes one of the effectively alternative pathways to circumvent the problem [10]. In particular, bromo-methyl phenyl sulfone, a readily accessible, stable, easily handled reagent, has been successfully introduced for C–H functionaliza-tion. The benzenesulfonyl (PhSO2-) group could be easily removed to reveal a methyl group by reduction reaction, providing easy access to methylated compounds. In 2014, Baran and co-workers [11] reported an elegant C–H methylation of heteroarenes. A masked zinc sulfinate reagent was designed and synthesized from bromomethyl phenyl sulfone. Afterwards, bromomethyl phenyl sulfone was reported to directly generate phenylsulfonyl methyl radical for introducing a methyl group through a two-step synthetic procedure [12].
In the past few years, much attention has been drawn on the imidazopyridine and their derivatives due to their unique bioactivity [13] and chemical properties [14]. There already are several marketed drugs containing the imidazo[1, 2-a]pyridine unit, including zolpidem (hypnotic) [15], alpidem (anxiolytic) [16] and olprinone (PDE-3 inhibitor) [17]. As a consequence, represen-tative synthetic approaches have been developed for the function-alization of imidazoheterocycles [18]. In comparison, the synthesis of imidazo[1, 2-a]pyridines bearing a sulfonylmethyl substituent at C-3 has been rarely reported [19]. Recently, visible light-induced photoredox catalysis has emerged as a powerful technique in organic synthesis because of its environmental compatibility, excellent functional group tolerance, high reactivity and versatility [20]. As a part of our continued studies focusing on clean C–H functionalization [21], herein we disclose an efficient and mild protocol for sulfonylmethylation of imidazopyridines using Ir(ppy)3 as a photo catalyst at room temperature through a visible-light irradiated radical process, providing an attractive approach to various sulfonylmethylated imidazopyridines. The products could be conveniently transformed into methylated compounds through reduction.
Our initial work started from the reaction of imidazo[1, 2-a] pyridine (1a) with bromomethyl phenyl sulfone (2a) in the presence of 2 mol% Ir(ppy)3 in MeCN. It was encouraging to see that the desired product 3aa was obtained in 46% yield after 12 h of irradiation (26 W CFL) at room temperature (Table 1, entry 1). An improved yield of 78% was achieved using 2 equiv. of Li2CO3 as base (Table 1, entry 2). Other solvents, such as DMSO (dimethyl sulfoxide), 1, 2-DCE (1, 2-dichloroethane), toluene, THF (tetrahydro-furan) were screened and MeCN proved to be particularly effective (Table 1, entries 3–6). Subsequent screening base, including Et3N, K2CO3, NaOAc, NaOH and K2HPO4, revealed that the yield was further increased to 89% with K2HPO4 (Table 1, entries 7–11). Moreover, the yield of the desired product decreased using other photo catalysts, such as Ru(bpy)3Cl2 and eosin Y (Table 1, entries 12 and 13). Shortening the reaction time to 8 h resulted in a lower yield of 72% yield (Table 1, entry 14). Finally, the control experiments suggested that photo-catalyst and light were essen-tial for this reaction (Table 1, entries 15 and 16).
|
|
Table 1 Optimization of the reaction conditions.a |

{kind=link}
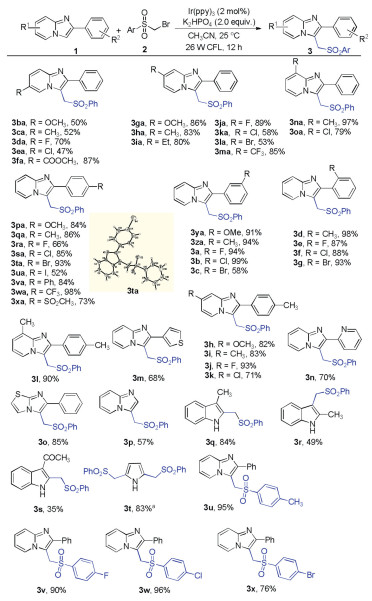
With the optimized conditions in hand (Table 1, entry 11), the generality and scope of the substrates were explored (Scheme 1). Initially, substrates with substituents on the C6, C7, and C8 positions of 2-phenylimidazo[1, 2-a]pyridines were examined. Various functional groups, including methoxy, methyl, ethyl, halogen and ester, were well tolerated under optimized reaction conditions (3ba-3oa). Moderate to excellent yields were obtained for most cases. While weak electron-withdrawing group (Cl) at C6 position led to a low yield (3ea). Furthermore, the effect of the substituent on the C2 phenyl ring was examined. Imidazo[1, 2-a]pyridines bearing electron-donating groups (OMe, Me, Ph) as well as electron-withdrawing groups (halogen, CF3, SO2CH3) on the 2-phenyl ring efficiently reacted with 2a to produce the C-3 sulfonylmethylated imidazo[1, 2-a]pyridine derivatives 3pa-3g in 52%–99% yields. The structure of 3ta was confirmed by single crystal X-ray crystallo-graphic analysis (CCDD No: 1903608). It is worth mentioning that steric hindrance could not be observed for congested o-substituted imidazo[1, 2-a]pyridines (3d-3g). In addition, di-substituted imi-dazo[1, 2-a]pyridines, with substitutions on both the pyridine and C2-phenyl ring also provided the corresponding products 3h-3l in high yields (71%–93%). 2-Thienyl and 2-pyridyl-substituted imidazo [1, 2-a]pyridine gave the products 3 m and 3n in 68% and 70% yields. Imidazo[2, 1-b]thiazole was also subjected to this reaction and produced the desired product 3o in good yield. Particularly, unsubstituted imidazo[1, 2-a]pyridine could be sulfonylmethylated, giving the target product 3p in 57% yield. Other heterocycles generally gave moderate to good yields of product in the case of indoles and pyrroles (3q-3t). Notably, this reaction tolerates free N H bonds and this method allows for the easy separation of product from starting material. To further examine the scope of the sulfonylmethylation reaction, the bromomethyl aryl sulfones were tested. Substituents, such as CH3, F, Cl, Br, were well tolerated and gave good to excellent yields (3u-3x).

|
Download:
|
| Scheme 1. Substrate scope. Reaction conditions: 1 (0.2 mmol), 2 (0.4 mmol), Ir(ppy)3 (2 mol%), K2HPO4 (0.4 mmol), CH3CN (1 mL) under N2 atmosphere at 25 ℃ for 12 h and at 26 W CFL irradiation. Isolated yields. a 4.0 equiv. of 2 for 24 h. | |
{kind=link}
Desulfonylation of 3aa, 3na, 3wa, 3a, 3q was easily achieved under reducing conditions (SmI2 in THF/H2O) to unveil the methyl group and afforded the target products 4aa, 4na, 4wa, 4a, 4q, respectively (Scheme 2).

|
Download:
|
| Scheme 2. Desulfonylation reveals the methyl group. | |
{kind=link}
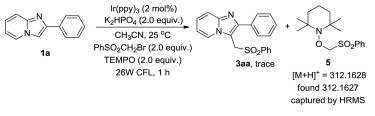
To understand the mechanism of this transformation, we attempted to perform the reaction under standard conditions with the addition of 2.0 equiv. of TEMPO (2, 2, 6, 6-tetramethylpiper-idinooxyl) as a radical scavenger (Scheme 3). Trace amount of product was obtained. However, the coupling product 5 of TEMPO with PhSO2CH2Br was captured by HRMS. These results indicated that the radical process might be involved in this sulfonylmethy-lation reaction.

|
Download:
|
| Scheme 3. Mechanism study. | |
{kind=link}
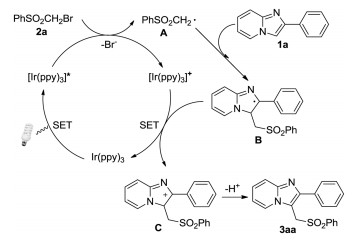
Based on these observations and literatures [11, 12], a possible catalytic cycle is proposed, as shown in Scheme 4. Firstly, the excited state [Ir(ppy)3*] was formed under light irradiation, which was next oxidized by PhSO2CH2Br 2a to generate a [Ir(ppy)3]+ complex and a ·CH2SO2Ph radical A. Subsequently, the radical A reacted with imidazo[1, 2-a]pyridine 1a to produce the intermediate B. Then, intermediate B was oxidized by [Ir(ppy)3]+ to form the intermediate C along with the regeneration of Ir(ppy)3. Finally, the deprotonation of C assisted by base afforded the desired product 3aa.

|
Download:
|
| Scheme 4. Proposed mechanism. | |
{kind=link}
In summary, we have successfully developed an efficient visible-light-induced C3-H sulfonylmethylation of imidazopyri-dines with bromomethyl aryl sulfones using Ir(ppy)3 as a photo-catalyst. Various sulfonylmethylated imidazopyridines were prepared in moderate to excellent yields with a wide range of functional group tolerance at room temperature. Meanwhile, desulfonylation of several selected compounds was easily achieved under reducing conditions to unveil the methyl group. The method may be an attractive strategy to install a methyl group on imidazopyridines.
Declarationof competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work was financially supported by the National Natural Science Foundation of China (No. 21602046), the MOST of China (No. 2016YFE0132600) and the Doctor's Scientific Research Foundation of Henan University of Chinese Medicine (No. BSJJ2016-12).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.09.040.
| [1] |
(a) P.A. Jones, D. Takai, Science 293 (2001) 1068-1070; (b) Y. Umezawa, O. Nishio, Nucleic Acids Res. 30 (2002) 2183-2192; (c) R. Jaenisch, A. Bird, Nat. Genet. 33 (2003) 245-254; (d) H. Sasaki, Y. Matsui, Nat. Rev. Genet. 9 (2008) 129-140; (e) T.V. Mishanina, E.M. Koehn, A. Kohen, Bioorg. Chem. 43 (2012) 37-43; (f) L. Zhang, X. Ding, J. Cui, et al., Nature 481 (2012) 204-208. |
| [2] |
(a) E.J. Barreiro, A.E. Kummerle, C.A. Fraga, Chem. Rev. 111 (2011) 5215-5246; (b) H. Schçnherr, T. Cernak, Angew. Chem. Int. Ed. 52 (2013) 12256-12267. |
| [3] |
F. Serpier, F. Pan, W.S. Ham, et al., Angew. Chem. Int. Ed. 57 (2018) 10697-10701. DOI:10.1002/anie.201804628 |
| [4] |
(a) Y. Chen, Chem. -Eur. J. 25 (2019) 3405-3439; (b) G. Yan, A.J. Borah, L. Wang, M. Yang, Adv. Syn. Catal. 357 (2015) 1333-1350. |
| [5] |
(a) F. Minisci, R. Bernardi, F. Bertini, R. Galli, M. Perchinummo, Tetrahedron 27 (1971) 3575-3579; (b) C. Punta, F. Minisci, Trends Heterocycl. Commun. Chem. 13 (2008) 1-68; (c) M.A. Duncton, Med. Chem. Res. 2 (2011) 1135-1161; (d) J. Tauber, D. Imbr, T. Opatz, Molecules 19 (2014) 16190-16222. |
| [6] |
(a) M. Janda, J. Šrogl, I. Stibor, M. Němec, P. Vopatrna', Tetrahedron Lett. 14 (1973) 637-638; (b) M. Araki, M. Maeda, Y. Kawazoe, Tetrahedron 32 (1976) 337-340; (c) D.A. DiRocco, K. Dykstra, S. Krska, et al., Angew. Chem. Int. Ed. 53 (2014) 4802-4806; (d) P.Z. Zhang, J.A. Li, L. Zhang, et al., Green Chem. 19 (2017) 919-923. |
| [7] |
(a) B.M. Bertilsson, B. Gustafsson, I. Kühn, K. Torssell, Acta Chem. Scand. 24 (1970) 3590-3598; (b) C. Giordano, F. Minisci, V. Tortelli, E. Vismara, J. Chem. Soc., Perkin Trans. Ⅱ 2 (1984) 293-295; (c) C. Crean, N.E. Geacintov, V. Shafirovich, J. Phys. Chem. B 113 (2009) 12773-12781; (d) K. Kawai, Y.S. Li, M.F. Song, H. Kasai, Bioorg. Med. Chem. Lett. 20 (2010) 260-265. |
| [8] |
(a) J. Jin, D.W.C. Macmillan, Nature 525 (2015) 87-90; (b) K. Natte, H. Neumann, M. Beller, R.V. Jagadeesh, Angew. Chem. Int. Ed. 56 (2017) 6384-6394; (c) W. Liu, X. Yang, Z.Z. Zhou, C.J. Li, Chem 2 (2017) 688-702. |
| [9] |
(a) M. Maeda, K. Nushi, Y. Kawazoe, Tetrahedron 30 (1974) 2677-2682; (b) S. Hix, M.S. Morais, O. Augusto, Free Radical Biol. Med.19 (1995) 293-301; (c) W. Jo, J. Kim, S. Choi, S.H. Cho, Angew. Chem. Int. Ed. 55 (2016) 9690-9694. |
| [10] |
(a) A. Gualandi, E. Matteucci, F. Monti, et al., Chem. Sci. 8 (2017) 1613-1620; (b) Q. Huang, S.Z. Zard, Org. Lett. 20 (2018) 1413-1416; (c) S.C. Lu, H.S. Li, Y.L. Gong, et al., J. Org. Chem. 83 (2018) 15415-15425.
|
| [11] |
J. Gui, Q. Zhou, C. Pan, et al., J. Am. Chem. Soc. 136 (2014) 4853-4856. DOI:10.1021/ja5007838 |
| [12] |
(a) F. Liu, P. Li, J. Org. Chem. 81 (2016) 6972-6979; (b) G. Filippini, M. Silvi, P. Melchiorre, Angew. Chem. Int. Ed. 56 (2017) 4447-4451. |
| [13] |
(a) A.T. Baviskar, S.M. Amrutkar, N. Trivedi, et al., ACS Med. Chem. Lett. 6 (2015) 481-485; (b) C. Enguehard-Gueiffier, A. Gueiffier, Mini-Rev. Med. Chem. 7 (2007) 888-899. |
| [14] |
(a) A. Douhal, F. Amat-Guerri, A.U. Acuña, J. Phys. Chem. 99 (1995) 76-80; (b) S. Takizawa, J. Nishida, Y. Yamashita, Mol. Cryst. Liq. Cryst. 455 (2006) 381-385. |
| [15] |
T.S. Harrison, G.M. Keating, CNS Drugs 19 (2005) 65. DOI:10.2165/00023210-200519010-00008 |
| [16] |
S.Z. Langer, S. Arbilla, J. Benavides, B. Scatton, Adv. Biochem. Psychopharmacol. 46 (1990) 61-72. |
| [17] |
T. Ueda, K. Mizusgige, K. Yukiiri, T. Takahashi, Cerebrovasc. Dis. 16 (2003) 396-401. DOI:10.1159/000072563 |
| [18] |
(a) J. Koubachi, S. El Kazzouli, M. Bousmina, G. Guillaumet, Eur. J. Org. Chem. (2014) 5119-5138; (b) A.K. Bagdi, A. Hajra, Chem. Rec. 16 (2016) 1868-1885; (c) C. Ravi, S. Adimurthy, Chem. Rec. 17 (2017) 1019-1038; (d) L. Tian, S. Lu, Z. Zhang, et al., J. Org. Chem. 84 (2019) 5213-5221. |
| [19] |
S. Lu, X. Zhu, K. Li, et al., J. Org. Chem. 81 (2016) 8370-8377. DOI:10.1021/acs.joc.6b01552 |
| [20] |
(a) C.K. Prier, D.A. Rankic, D.W.C. MacMillan, Chem. Rev.113 (2013) 5322-5363; (b) D. Staveness, I. Bosque, C.R.J. Stephenson, Acc. Chem. Res. 49 (2016) 2295-2306; (c) C.S. Wang, P.H. Dixneuf, J.F. Soule', Chem. Rev. 118 (2018) 7532-7585; (d) S. Crespi, M. Fagnoni, Top. Heterocycl. Chem. 54 (2018) 1-70; (e) L. Marzo, S.K. Pagire, O. Reiser, B. König, Angew. Chem. Int. Ed. 57 (2018) 10034-10072. |
| [21] |
(a) J. Wu, X. Cui, L. Chen, G. Jiang, Y. Wu, J. Am. Chem. Soc. 131 (2009) 13888-13889; (b) Z. Wu, C. Pi, X. Cui, J. Bai, Y. Wu, Adv. Synth. Catal. 355 (2013) 1971-1976; (c) C. Pi, X. Cui, X. Liu, et al., Org. Lett. 16 (2014) 5164-5167; (d) C. You, C. Pi, Y. Wu, X. Cui, Adv. Synth. Catal. 360 (2018) 4068-4072; (e) L. Wang, D. Xiong, L. Jie, C. Yu, X. Cui, Chin. Chem. Lett. 29 (2018) 907-910; (f) Z. Yang, L. Jie, Z. Yao, Z. Yang, X. Cui, Adv. Synth. Catal. 361 (2019) 214-218; (g) T. Yuan, C. Pi, C. You, et al., Chem. Commun. 55 (2019) 163-166; (h) C. Pi, X. Yin, X. Cui, Y. Ma, Y. Wu, Org. Lett. 21 (2019) 2081-2084; (i) J. Ren, C. Pi, Y. Wu, X. Cui, Org. Lett. 21 (2019) 4067-4071; (j) Z. Shen, C. Pi, X. Cui, Y. Wu, Chin. Chem. Lett. 30 (2019) 1374-1378. |