 2019, Vol. 30
2019, Vol. 30 


b Key Laboratory of Polymer Ecomaterials, Changchun Institute of Applied Chemistry, Chinese Academy of Science, Changchun, China
Polypeptides containing α-amino acids possess fascinating diverse structures, displaying significant similarities to the complex secondary structure of natural biomolecules [1, 2]. This natural similarity has spurred various strategies based on synthetic polypeptides, such as self-assembly of block copolymer and organic/inorganic multi-layer composite, to devise and fabricate smart materials, especially soft and nanostructured hybrid materials, for bone repair engineering [3, 4], bio-sensing [5], and drug delivery [6-8]. In the stepwise fabrication of soft matters, the primary step involves self-assembly of amphiphilic copolymers, more recently derived from biocompatible or biodegradable blocks, into vesicles, frequently as so-called polymersomes [9, 10]. Peptides or drugs are finally loaded in polymersomes in physical encapsulation manner by taking advantage of the surface charges [11, 12]. However, these approaches failed to realize controlled dissociation or triggered drug release, which is of great importance in enhancing the system's anticancer efficacy thus minimizing its side effects to healthy cells and organs [13, 14]. Nanomaterials, organic-inorganic nanohybrids in particular, which can be used as stimuli-responsive materials, are attracting more and more attention in nanomedicine and bio-related fields [15-18]. Among various nanomaterials [19-22], mesoporous silica nanoparticles (MSNs) stand out owing to their tunable pore diameters, excellent biocompatibility, and superior stability [23, 24]. Although abundant materials have been designed as switchable gating entities to construct smart MSN-based antican-cer nanosystems [25-30], very limited MSN-based platforms have been installed and regulated with biomolecules-based gates for controlled drug release. Combinatorial hybrid materials based on the biocompatible polypeptide-modified MSNs possesses supple-mentary merits since they not only have large loading capacity and good stability but also exhibit excellent biocompatibility [31-33]. The incorporation of tunable switches in the fabrication of drug nanocarriers provides a new possibility to achieve precise drug release at targeted lesion locations, thus improving the anticancer efficiency [34, 35]. Moreover, it should be noted that receptor-targeted therapeutic nanosystems based on inorganic-organic composites are emerging as promising candidates to foster the progress of improving cancer inhibition capability [36-38].
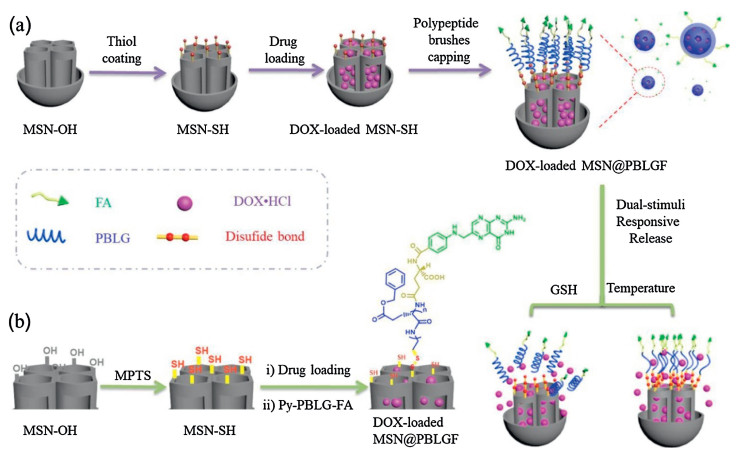
In this proof-of-concept study, we fabricate a hydrophobic poly(γ-benzyl-L-glutamate) (PBLG)-based synthetic polypeptide brushes-functionalized MSN as a new stimuli-responsive polymer-inorganic hybrid drug delivery platform, namely MSN@PBLGF (Fig. 1). Hydrophilic folic acid (FA) has been employed as a primary targeted group to be covalently attached on one end of PBLG before its connection onto MSN surface.

|
Download:
|
| Fig. 1. Schematic representation of the preparation process of MSN@PBLGF nanoplatform and its operation in response to GSH and temperature dual-stimuli for targeted drug delivery. | |
{kind=link}
Since the existence of PBLG, the DOX-loaded MSN@PBLGF system could release drugs at higher temperature due to the conformation transition from rodlike α-helix conformation to a random coil conformation [35, 39]. Besides, disulfide linkage between MSNs and PBLG can be destroyed by GSH, thus losing PBLG brushes and allowing drug release. Furthermore, FA in the system can not only provide the tumor-targeted function but also greatly improve the biocompatibility of this system.
Hydroxyl-modified MSN (MSN-OH) was prepared through traditional hydrothermal method, followed by further post-modification via silanization to introduce -SH groups (Fig. 1). Then, the resulting MSN-SH nanoparticles were immersed in the aqueous solution of DOX to load drugs into the MSN pores via concentration gradient. For the capping of MSN with PBLG polypeptide brushes, the newly synthesized Py-PBLG-FA (see the Supporting information) was reacted with MSN-SH through thiol-disulfide exchange reaction to result in DOX-loaded MSN@PBLGF. The symmetrical spherical morphology of the constructed hybrid nanomaterial was clearly visualized by scanning electron micros-copy (SEM) (Figs. 2a and b). The average diameter (about 74 nm) of the MSN@PBLGF nanoparticles was larger than that of MSN-OH (ca. 65 nm), attributed to the surface-modification of PBLGF biopolymer brushes. Transmission electron microscopy (TEM) images (Figs. 2c and d) showed the spherical morphology of bare MSN-OH with the typical mesoporous character of MSN. However, after successful modification of MSNs with PBLGF, the repeated channels were partly sheltered by those polypeptide brushes thus causing the relatively rough surface of composite nanoparticles. Moreover, the hydrodynamic diameters of relevant nanoparticles were measured by dynamic light scattering (DLS). As shown in Fig. S11a (Supporting information), the hydrodynamic diameter of MSN-OH was measured to be 164 nm in average, and the decoration of PBLGF on MSN-OH significantly increased the hydrodynamic diameter of the particles (ca. 250 nm), larger than those calculated from TEM analysis. This discrepancy in hydrody-namic particle sizes can be explained by two factors: 1) the formation of a hydrate layer around the nanoparticles in aqueous solution; 2) the occurrence of aggregation in test dispersant system [40]. And the potentials were also tested, as shown in Fig. S11b (Supporting information). Generally, the absolute values of potential less than 30 mV imply that the formation of aggregates will occur due to the reduced electrostatic repulsion among the particles. Meanwhile, nanoparticles with an absolute value of zeta potential larger than 30 mV have been regarded as a stable system [41]. This further verified the MSN-OH with a negative value (-10.4 mV) tend to form aggregates, in consistency with the data of hydrodynamic diameter. After functionalization with PBLGF, however, composite nanoparticles exhibited more negatively charged surface (-29.4 mV), which not only suggested the successful decoration of carboxyl-containing polymers but also implied the formation of a stable suspension in distilled water.

|
Download:
|
| Fig. 2. SEM images of (a) MSN-OH and (b) MSN@PBLGF. TEM images of (c) MSN-OH and (d) MSN@PBLGF. | |
{kind=link}
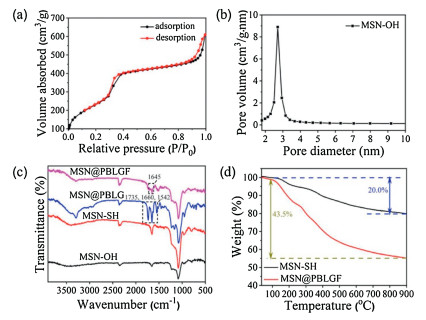
N2 adsorption/desorption isotherms of MSN-OH showed a type IV curve, suggesting hysteresis characteristics of mesoporous structure. The BET surface areas and BJH average pore size were 908 m2/g (Fig. 3a) and 2.71 nm (Fig. 3b), respectively. The grafting process of the pore structure can be further confirmed by low angle powder X-ray diffraction (PXRD) (Fig. S12 in Supporting informa-tion). The crystalline structure of the resultant material was still maintained although the intensity of the diffraction peaks slightly decreased after the functionalization of biopolymer brushes. Fourier-transform infrared (FT-IR) spectroscopy was used to monitor the progress of modification (Fig. 3c). The presence of representative absorption signals of MSN-based materials at 1086 and 801 cm-1 certified the successful construction of bare mesoporous silica nanoparticle. After modification with PBLGF, the prominent peaks appeared at 1735 and 1542 cm-1 corre-sponding to -NH-C=O stretching and bending, respectively, demonstrated the successful coating of PBLG brushes on MSNs, and the characteristic variations at 1645 cm-1 could be assigned to the existence of folic acid. Thermogravimetric analysis (TGA) indicated a moderate decline of MSN-SH and MSN@PBLGF below 200 ℃, where the absorbed water began to vaporize, and the TGA of MSN@PBLGF demonstrated that PBLGF entities accounted for ca. 23.5% of mass loss between 160 and 900 ℃ (Fig. 3d). These data proved the successful grafting and capping of MSN with PBLGF brushes.

|
Download:
|
| Fig. 3. (a) N2 adsorption-desorption isotherms of MSN-OH. (b) Pore size distribution of MSN-OH. (c) FT-IR spectra of MSN-OH, MSN-SH, MSN@PBLG]FID$T51[and MSN@PBLGF. (d) TGA curves of MSN-SH and MSN@PBLGF. | |
{kind=link}
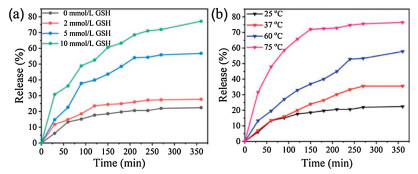
Illuminated by the abnormal GSH concentration (2–10 mmol/L) of cancer intracellular microenvironment, which is greatly deviated from the normal level (CGSH=2–20 μmol/L), GSH stimu-lus-responsive drug release proves an efficient modality of targeted cancer chemotherapy. To verify GSH controlled release performance of as-synthesized MSNs material, the release profiles of DOX-loaded MSN@PBLGF were recorded with different GSH concentrations (0, 2, 5 and 10 mmol/L). As in Fig. 4a, without GSH addition, only ca. 19% DOX was released from the material. However, the release of DOX progressed upon elevating the GSH concentration, that is, higher concentration of GSH could break the disulfide bonds efficiently, resulting in the fall-off of biopolymers followed by the rapid escape of DOX from the pores. Consequently, the release amount of DOX at 10 mM GSH performed around four times higher than that in the absence of GSH in the 360 min period.

|
Download:
|
| Fig. 4. The release profiles of the DOX-loaded MSN@PBLGF under various external stimuli: (a) GSH, and (b) temperature. | |
{kind=link}
Furthermore, PBLG brushes with the inherent thermal-respon-sive feature endowed the DOX-loaded nanosystem with the capability of temperature responsiveness [39]. To clarify the thermal sensibility of PBLG, the release experiments of DOX-loaded MSN@PBLGF were carried out at different temperatures (25, 37, 60 and 75 ℃). As in Fig. 4b, temperature increase led to the acceleration of drug release due to the weakening of hydrogen-bonding interactions of amine acid residues. The drugs could be encapsulated steadily at room temperature with an acceptable premature release (only 19% of the drug within 6 h), while the elevated temperature changed the confirmation of PBLG brushes on the surface of MSN, causing the unblocking of MSN mesopores. When the temperature reached 75 ℃, the DOX release was significantly accelerated, and almost 75% of the encapsulated DOX was released within 6 h. All of the above proof-of-concept experimental data proved that DOX-loaded MSN@PBLGF nanosystem is capable of GSH/ temperature dual-responsive release in tumor microenvironment.
In order to verify whether the FA entities attached on the outermost surface of MSN@PBLGF nanoplatform could guide these nanoparticles preferentially to cancer cells, experiments on A549 cells were performed with fluorescent microscopy, in which the cells treated with MSN@PBLG (without the FA segment) were used as the control. Free DOX, DOX-loaded MSN@PBLG nanoparticles, and DOX-loaded MSN@PBLGF nanomaterials were incubated with folate receptors (FR) over-expressing A549 cells, respectively. As shown in Fig. 5a (left), after culturing for 2 h, the free DOX group showed weak intracellular red fluorescence, while the DOX-loaded MSN@PBLGF nanosystem group showed a little stronger red fluorescence under the same conditions. However, associated with extending the incubation time to 4 h, the DOX-loaded MSN@PBLGF group showed much stronger red fluorescence than the free DOX group, and the DOX-loaded MSN@PBLG group lacking of FR entities still showed weak red fluorescence, which demonstrated that MSN@PBLGF could be ingested more efficiently due to the folate receptor-mediated endocytosis in a time-dependent manner. The intracellular fluorescence clearly indicated that the increasing concentration of DOX from DOX-loaded MSN@PBLGF internalized into the cells with extended culture time. Significantly, the red fluorescence of DOX was mainly accumulated in the cell nucleus, suggesting that the released DOX from these nanoparticles successfully entered into the cell nuclei.

|
Download:
|
| Fig. 5. (a) Fluorescence images of A549 cells and LO2 cells incubated with free DOX, DOX-loaded MSN@PBLG, and DOX-loaded MSN@PBLGF for 2 h and 4 h, respectively. Blue represents nuclei stained with DAPI and red represents DOX (scale bar: 50 mm). Note: for detail, see Fig. S14 in Supporting information. (b) Cytotoxicity assay of A549 cells in the presence of DOX, non-loaded MSN@PBLGF, and DOX-loaded MSN@PBLGFcultured for 24 husing CCK-8 assay. | |
{kind=link}
To verify the biocompatibility of MSN@PBLGF nanosystem toward normal cells, intracellular DOX release from DOX-loaded MSN@PBLGF in human hepatocyte cells (LO2) was further investigated using fluorescence microscopy. As shown in Fig. 5a (right), small amount of DOX could be witnessed in the cells during the 4 h in DOX-loaded MSN@PBLGF group, which is most likely due to the low expression of folate receptor in the membrane of normal cells, while significant DOX fluorescence was observed in the nucleus of cells incubating with free DOX. Moreover, upon culturing with DOX-loaded MSN@PBLG for 4 h, the subcellular distribution of DOX suggested more compared with the case of DOX-loaded MSN@PBLGF, further confirming the recognition capability of the folate entity. These experiments demonstrated that the FA group attached on the surface of DOX-loaded MSN@PBLGF extensively enhanced the specific targeting of smart nanosystems towards tumor cells, effectively reducing the side effects and severe damage of anticancer drugs to normal cells and organs.
Meanwhile, extended experiments of assessing anti-cancer efficiency were performed by CCK-8 assay using A549 cell line. Free DOX, DOX-loaded MSN@PBLG, and DOX-loaded MSN@PBLGF were cultured with A549 cells for 24 h. Fig. 5b demonstrated that, compared with the free DOX group and DOX-loaded MSN@PBLGF group, the assay results of DOX non-loaded MSN@PBLGF group demonstrated high cell viabilities of up to 94% even reaching to 50 mg/mL, indicating the good biocompatibility of MSN@PBLGF and great potential of this nanoplatform in biological applications. Meanwhile, DOX-loaded MSN@PBLGF nanosystem displayed comparable anticancer activity against A549 cells compared with free DOX with the increased amount of drug. The excellent biocompatibility of MSN@PBLGF endowed it great potential to serve as a desirable cancer theranostic nanoplatform.
In summary, we constructed a new synthetic polypeptide brushes-coated organic-inorganic nanohybrid drug delivery sys-tem (MSN@PBLGF) and achieved its targeted and controlled drug release in response to GSH and thermal stimuli. PBLG polymers modified on MSN surfaces through disulfide bonds could not only be dissociated by increasing the concentration of GSH but also transform from rod-like α-helical structure to random conforma-tion in response to an elevated temperature. In addition, the incorporation of FA entities makes the hybrid nanosystem recognizable specifically by cancer cells. Furthermore, the MSN@PBLGF nanoplatform showed excellent biocompatibility while DOX-loaded MSN@PBLGF system exhibited good cancer cell inhibition performance, as illustrated by in vitro cell-growth inhibition assay. We envision that this drug delivery system will become useful in effective targeted tumor therapy and could expand the pools of hybrid medical nanomaterials.
AcknowledgmentWe greatly acknowledge the Jilin Province-University Coopera-tive Construction Project-Special Funds for New Materials (No. SXGJSF2017-3) for financial support.
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.08.017.
| [1] |
F.E. Karasz, G.E. Gajnos, Biopolymers 15 (1976) 1939-1950. DOI:10.1002/bip.1976.360151006 |
| [2] |
F. Chécot, S. Lecommandoux, Y. Gnanou, H.A. Klok, Angew. Chem. Int. Ed. 41 (2002) 1339-1343. DOI:10.1002/1521-3773(20020415)41:8<1339::AID-ANIE1339>3.0.CO;2-N |
| [3] |
M. Liong, J. Lu, M. Kovochich, et al., ACS Nano 2 (2008) 889-896. DOI:10.1021/nn800072t |
| [4] |
J.E. Lee, N. Lee, T. Kim, J. Kim, T. Hyeon, Acc. Chem. Res. 44 (2011) 893-902. DOI:10.1021/ar2000259 |
| [5] |
K. Ariga, Q. Ji, T. Mori, et al., Chem. Soc. Rev. 42 (2013) 6322-6345. DOI:10.1039/c2cs35475f |
| [6] |
J.L. Vivero-Escoto, I.I. Slowing, B.G. Trewyn, V.S. Lin, Small 6 (2010) 1952-1967. DOI:10.1002/smll.200901789 |
| [7] |
Y.W. Yang, Med. Chem. Commun. 2 (2011) 1033-1049. DOI:10.1039/c1md00158b |
| [8] |
P. Yang, S. Gai, J. Lin, Chem. Soc. Rev. 41 (2012) 3679-3698. DOI:10.1039/c2cs15308d |
| [9] |
B.M. Discher, Y.Y. Won, D.S. Ege, et al., Science 284 (1999) 1143-1146. DOI:10.1126/science.284.5417.1143 |
| [10] |
D.E. Discher, A. Eisenberg, Science 297 (2002) 967-973. DOI:10.1126/science.1074972 |
| [11] |
M. Kar, N. Tiwari, M. Tiwari, M. Lahiri, S.S. Gupta, Part. Part. Syst. Charact. 30 (2013) 166-179. DOI:10.1002/ppsc.201200089 |
| [12] |
R. Liu, X. Zhao, T. Wu, P. Feng, J. Am. Chem. Soc. 130 (2008) 14418-14419. DOI:10.1021/ja8060886 |
| [13] |
C. Widakowich, G. de Castro Jr, E. de Azambuja, P. Dinh, A. Awada, Oncologist 12 (2007) 1443-1455. DOI:10.1634/theoncologist.12-12-1443 |
| [14] |
M. Hoop, A.S. Ribeiro, D. Rösch, et al., Adv. Funct. Mater. 28 (2018) 1705920. DOI:10.1002/adfm.201705920 |
| [15] |
Z. Li, N. Song, Y.W. Yang, Matter 1 (2019) 345-368. DOI:10.1016/j.matt.2019.05.019 |
| [16] |
T.M. Guardado-Alvarez, L. Sudha Devi, M.M. Russell, B.J. Schwartz, J.I. Zink, J. Am. Chem. Soc. 135 (2013) 14000-14003. DOI:10.1021/ja407331n |
| [17] |
N. Song, X.Y. Lou, L. Ma, et al., Theranostics 9 (2019) 3075-3093. DOI:10.7150/thno.31858 |
| [18] |
N.K. Mal, M. Fujiwara, Y. Tanaka, Nature 421 (2003) 350. DOI:10.1038/nature01362 |
| [19] |
X. Li, J. Han, X. Wang, et al., Mater. Chem. Front. 3 (2019) 103-110. DOI:10.1039/C8QM00509E |
| [20] |
L.L. Tan, N. Song, S.X.A. Zhang, et al., J. Mater. Chem. B 4 (2016) 135-140. DOI:10.1039/C5TB01789K |
| [21] |
S. Li, B.A. Moosa, J.G. Croissant, N.M. Khashab, Angew. Chem. Int. Ed. 54 (2015) 6804-6808. DOI:10.1002/anie.201501615 |
| [22] |
Z. Zhang, W. Cao, H. Jin, et al., Angew. Chem. Int. Ed. 48 (2009) 9171-9175. DOI:10.1002/anie.200903112 |
| [23] |
C. Argyo, V. Weiss, C. Bräuchle, T. Bein, Chem. Mater. 26 (2013) 435-451. |
| [24] |
R.K. Singh, K.D. Patel, K.W. Leong, H.W. Kim, ACS Appl. Mater. Interfaces 9 (2017) 10309-10337. |
| [25] |
C.Y. Lai, B.G. Trewyn, D.M. Jeftinija, et al., J. Am. Chem. Soc. 125 (2003) 4451-4459. DOI:10.1021/ja028650l |
| [26] |
S. Giri, B.G. Trewyn, M.P. Stellmaker, V.S.Y. Lin, Angew. Chem. Int. Ed. 44 (2005) 5038-5044. DOI:10.1002/anie.200501819 |
| [27] |
Y. Zhu, J. Shi, W. Shen, et al., Angew. Chem. Int. Ed. 44 (2005) 5083-5087. DOI:10.1002/anie.200501500 |
| [28] |
Q.L. Li, W.X. Gu, H. Gao, Y.W. Yang, Chem. Commun. (Camb.) 50 (2014) 13201-13215. DOI:10.1039/C4CC03036B |
| [29] |
N. Song, Y.W. Yang, Chem. Soc. Rev. 44 (2015) 3474-3504. DOI:10.1039/C5CS00243E |
| [30] |
Y. Wu, Y. Long, Q.L. Li, et al., ACS Appl. Mater. Interfaces 7 (2015) 17255-17263. DOI:10.1021/acsami.5b04216 |
| [31] |
J. Wang, J. Yu, Y. Yan, et al., Polym. Adv. Technol. 30 (2019) 1461-1472. DOI:10.1002/pat.4578 |
| [32] |
M. Yurdakul, H.A. Oktem, M.D. Yilmaz, ChemistrySelect 4 (2019) 2084-2088. DOI:10.1002/slct.201803783 |
| [33] |
Z. Zhang, H. Shi, Q. Wu, et al., New J. Chem. 43 (2019) 4581. DOI:10.1039/C8NJ05745A |
| [34] |
X.Y. Lou, Y.P. Li, Y.W. Yang, Biotechnol. J. 14 (2019) 1800354. DOI:10.1002/biot.201800354 |
| [35] |
Y. Masuda, T. Miyazawa, Die Makromol. Chemie 103 (1967) 261-267. DOI:10.1002/macp.1967.021030126 |
| [36] |
S. Santra, C. Kaittanis, O.J. Santiesteban, J.M. Perez, J. Am. Chem. Soc. 133 (2011) 16680-16688. DOI:10.1021/ja207463b |
| [37] |
S. Boonyarattanakalin, J. Hu, S.A. Dykstra-Rummel, A. August, B.R. Peterson, J. Am. Chem. Soc. 129 (2007) 268-269. DOI:10.1021/ja067674f |
| [38] |
T.T. Beaudette, J.A. Cohen, E.M. Bachelder, et al., J. Am. Chem. Soc. 131 (2009) 10360-10361. DOI:10.1021/ja903984s |
| [39] |
A. Kagemoto, R. Fujishiro, Die Makromol. Chemie 114 (1968) 139-145. DOI:10.1002/macp.1968.021140110 |
| [40] |
C.J. Sunderland, M. Steiert, J.E. Talmadge, A.M. Derfus, S.E. Barry, Drug Develop. Res. 67 (2006) 70-93. DOI:10.1002/ddr.20069 |
| [41] |
V.J. Mohanraj, Y. Chen, J. Trop, Pharm. Res. 5 (2006) 561-573. |