 2019, Vol. 30
2019, Vol. 30 


b Jiangxi Province Engineering Research Center of Ecological Chemical Industry, Jiujiang University, Jiujiang, China, br, c School of Pharmaceutical and Chemical Engineering, Taizhou University, Taizhou, China;
c School of Pharmaceutical and Chemical Engineering, Taizhou University, Taizhou, China
Imines, as Schiff bases, are well known as a very versatile intermediate that are widely applied in the production of pesticides, medicine, and so on [1, 2]. Conventional synthetic method of imines usually involves condensation reaction between amines and carbonyl compounds, and requires unstable aldehydes, Lewis acid catalysts, and dehydrating agents [1, 3], which restricted the practical use of this method [4].
The oxidative self-coupling of amines to imines was one of the most efficient methodologies [1]. However, most of the reported protocols required hazardous stoichiometric oxidants such as permanganate, chromate, and 2-iodoxybenzoic acid, which was undesirable from the perspective of green chemistry [3, 5]. Replacement of these undesired oxidants with green oxidants like air or O2 is an attractive and active research area. In the past decade, various heterogeneous catalysts have been applied to the self-coupling of primary amines, including catalysts based on Pt [6], Pd [7], Ru [8], Au [9], V [10], Cu [11], as well as other catalytic systems employing MOF [12], GO [13], and photocatalysts [14, 15], etc. However, despite the satisfying conversions obtained using some of the systems, most of them either required complicated synthetic procedure of catalysts, precious metal, toxic catalysts, harsh reaction conditions, light irradiation, prolonged reaction time, alkali addition, or suffered from poor reusability of the catalysts, and poor selectivity due to the formation of by-products. Therefore, effective heterogeneous catalytic system for the oxidative self-coupling of amines has long been required to overcome the problems mentioned above.
Among the available metal oxides, manganese oxides are cheap, non-toxic, and are effective for many catalytic processes, such as selective oxidation of various organic compounds [2, 16-22], CO oxidation [23], and water-splitting reaction [24]. Manganese oxide had superior catalytic activity due to the abundant active sites (Mn3+) it possesses, as well as excellent redox properties resulted from its multi-valency, weaker metal-oxygen bond it forms, and its rich lattice oxygen content with higher mobility [2, 17, 25].
Enhancing the selectivity of imines for the oxidative self-coupling of amines was found challenging [19, 26, 27]. High amine conversion was normally accompanied with the formation of by-products including nitrile, aldehyde, amide in the formation of imine [19]. Precipitation was one of the most favorite methods for catalyst preparation in large scale. Recently, Our latest research showed that Mn4+ in manganese oxides through simple oxalate route was responsible for the excessive oxidation of quinoline, while Mn3+ in manganese oxides displayed remarkable catalytic activity towards oxidative dehydrogenation of 1, 2, 3, 4-tetrahydroquinoline [28]. The valence distribution of manganese was a key factor to the high selectivity of quinoline. In this work, we prepared different manganese oxides by a simple oxalate route. The manganese oxalate calcined at a certain temperature (350 ℃, 450 ℃, 550 ℃) were denoted M-3, M-4, M-5. Detailed methods for catalyst preparation, characterization and evaluation were summarized in Supporting information. The structure and morphology of the catalyst were characterized by XRD, N2 sorption, SEM, TEM, and the results showed that M-3 was mesoporous Mn2O3 microrod with moderate surface area (Fig. 1, Table S1 and Figs. S1–S3 in Supporting information). As a novel attemptation for oxidation of benzylamine to imine, 100% conversion of benzylamine and 96.2% selectivity of imine were successfully achieved with M-3 catalyst in 2 h, which was far more superior than other existing protocols. Furthermore, this catalytic process was carried out under milder conditions, no base additives, and air as the only oxidant.

|
Download:
|
| Fig. 1. XRD patterns of wide angle (10°–80°) of the samples. | |
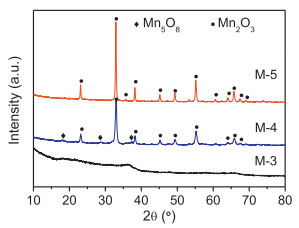
The X-ray diffraction patterns of the samples were showed in Fig. 1. M-3 exhibited no distinct diffraction peak in Fig. 1, which suggested the amorphous nature of the material. Sample M-4 showed clearly appeared diffraction peaks, which were consistent with the main cubic crystal Mn2O3 ((JCPDS Card No. 041-1442) and a small part of metastable Mn5O8 (JCPDS Card No. 039-1218). Sample M-5 only showed enhanced diffraction peaks of Mn2O3. The results implied that the crystal particle size of manganese oxides increased with calcination temperature elevated (Table S1).
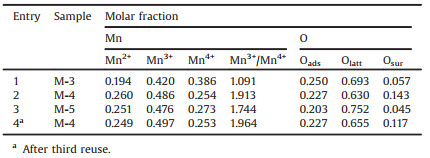
The surface elemental compositions were analyzed by using XPS. Fig. 2 displayed the Mn 2p and O 1s XPS spectra of various catalysts. It was well known that the deconvolution of Mn 2p3/2 and Mn 2p1/2 XPS spectra was useful to distinguish the oxidation states of Mnn+. As shown in Fig. 2a, the Mn 2p2/3 XPS spectra on the surface of catalysts showed a broad shoulder peak, which implied the coexistence of Mn2+, Mn3+ and Mn4+ species. The fitted Mn 2p3/2 peaks at the the binding energy (BE) of 643.0 eV, 641.7 eV, and 640.6 eV could be assigned to Mn4+, Mn3+, and Mn2+, respectively (Fig. 2a) [29, 30]. A spin-orbit energy separation (11.7 ± 0.2 eV) was displayed between Mn 2p1/2 and Mn 2p2/3 states (Fig. 2a). Fig. 2b showed the asymmetrical peak, which could be seen as the superposition of three peaks of O 1s at 533.0 eV, 531.3 eV, 529.8 eV. The small peak at BE of 533.0 eV was the characteristic of the molecular water (named as Osuf), the peak at BE of 531.3 eV corresponded to adsorbed oxygen species (O2-, O22-, O-, named as Oads), and the peak at BE of 529.8 eV was indexed to lattice oxygen atoms in Mn-O-Mn (O2-, named as Olatt) [29-31]. Table 1 showed surface Mnn+ and different surface oxygen species molar fraction from Mn 2p and O 1s XPS spectra. Surface-active Mn3+ species had an important influence on the catalytic activity of the oxidative self-coupling of amines to imines [19]. In our latest research, we had found that Mn4+ easily led to excessive oxidation of quinoline [28]. As could be interestingly seen from Table 1, M-4 possessed highest content of Mn3+ (0.486) and highest Mn3+/Mn4+ ratio, implying that M-4 possessed the moderate catalytic activity and higher selectivity of imines.Table 1 showedthat M-4 owned highest content of water in surface oxygen compared with the other samples. The role of water on manganese oxide surface was also believed to be significant for amine oxidation to imine [19].

|
Download:
|
| Fig. 2. Mn 2p and O 1s XPS spectra of various catalysts. | |
|
|
Table 1 Parameters of the fitted components on surface from Mn 2p and O 1s XPS spectra. |
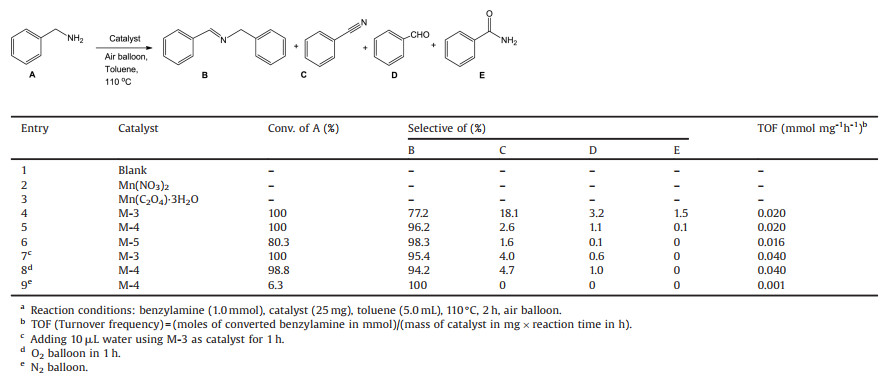
The catalytic activities of different manganese oxides were evaluated using the oxidative self-coupling of benzylamine as a model reaction. The evaluating results were shown in Table 2. The reaction hardly happened in the absence of catalyst (Table 2, entry 1). No products were detected using Mn(NO3)2, Mn(C2O4)·3H2O as the catalysts (Table 2, entries 2 and 3). Surprisingly, not only M-4 catalyst achieved 100% conversion of benzylamine, but also the selectivity of imine was as high as 96.2% (Table 2, entry 5). Concerning the solvent (Table S2 in Supporting information, entries 1–5), toluene solvent could provide higher conversion and selectivity. The by-product was mainly benzonitrile in toluene as the solvent, in addition to a small amount of benzaldehyde and benzamide. At 100% conversion of benzylamine, the selectivity to imine was only 77.2% by M-3 catalyst (Table 2, entry 4). When the conversion of benzylamine was 80.3%, the highest selectivity (98.3%) of imine was achieved using M-5 catalyst (Table 2, entry 6), implying that the imine showed higher selectivity at low conversion of benzylamine after 2 h of the reaction. These results suggested manganese oxide calcined at low temperature showed high catalytic activity for oxidation of amine, and manganese oxide calcined at high temperature showed high selectivity to imine. Although this reaction process produced water, the addition of trace water increased the selectivity of the imine (Table 2, entry 7, Fig. 3b). This suggested that trace water was beneficial to the formation of imines during the reaction, which was also consistent with the literature [19]. Under O2 atmosphere, M-4 achieved the enhanced conversion (98.8%) and the decreased selectivity (94.2%) after 1 h (Table 2, entry 8, Fig. 3a). N2 atmosphere only gave 6.3% conversion (Table 2, entry 9), which ascribed the oxidation role of active oxygen species in the manganese oxide [32].
|
|
Table 2 Imine synthesis from benzylamine over the as prepared catalysts.a |
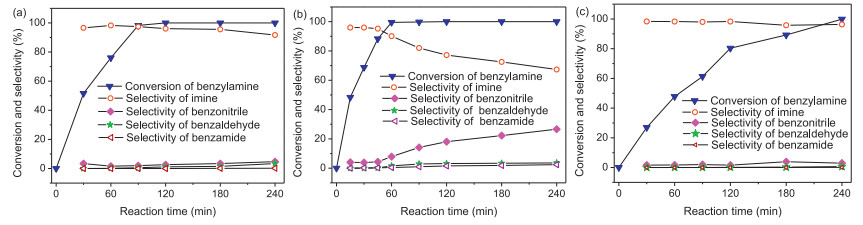
The selectivity of iminewas an important issue in the oxidation of benzylamine [19]. Figs. 3a–c showed catalytic activity of M-4, M-3, and M-5 dependent on the reaction time. M-4 gave 100% conversion of benzylamine and 96.2% selectivity of imine in 2 h, and the selectivity of imine slightly decreased when the reaction time was further prolonged (Fig. 3a). M-3 gave 99.5% conversion of benzyl-amine and 90.1% selectivity of imine for 1 h (Fig. 3b). The selectivity of imine sharply decreased by M-3 with the reaction time being prolonged, and the selectivity of by-product benzonitrile rapidly increased. Compared with M-3 and M-4, benzylamine conversion with M-5 catalyst significantly decreased at the same reaction time (Fig. 3c). M-5 gave only 80.3% conversion and 98.3% selectivity for 2 h, and the selectivity of imine also slightly decreased extending the reaction time to 4 h. These results further confirmed that M-4 was the best catalyst for the oxidation of benzylamine to imine. Testing of the different amount of catalyst (Table S3 in Supporting information), and different reaction temperatures (Table S4 in Supporting information) also confirmed that M-4 was the most effective catalyst. Compared with various manganese-based catalysts from the reported literature (Table S5 in Supporting information), undoped M-4 in our work not only achieved the higher conversion, but also gave the higher selectivity.

|
Download:
|
| Fig. 3. Catalytic activity of catalysts (a) M-4, (b) M-3, (c) M-5 dependent on the reaction time. Reaction conditions: benzylamine (1.0 mmol), catalyst (25 mg), toluene (5.0 mL), 110 ℃, air balloon. | |
The recyclability and heterogeneity of M-4 had been investi-gated by using benzylamine as the test substrate (Figs. S5–S8 in Supporting information). Each cycle gave good conversion and hardly significantly reduced selectivity (Fig. S5). Figs. S7 and S8 showed that the XRD pattern, Mn 2p XPS spectra of third reused M-4 displayed no significant change compared with that of the fresh catalyst. In addition, Mn3+/Mn4+ ratio did not significantly change in M-4 (Table 1, entries 2 and 4). To explore the leaching of active species, the hot filtration survey was also carried out. After the catalyst was removed at 76.0% conversion, the conversion of benzylamine had not been further improved (Fig. S6). It could be inferred from these results that M-4 was reusability, and authentically heterogeneous nature.
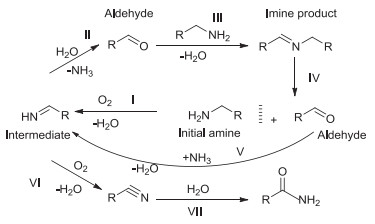
With the establishment of optimized conditions, the substrate scope and limitations were investigated using M-4 catalyst. The evaluation data of the oxidative self-coupling of a variety of amines, including aromatic, aliphatic amines, was showed in Table 3. M-4 was able to catalyze aromatic (Table 3, entries 1–5), aliphatic (Table 3, entries 6 and 7) amines to the corresponding imines. However, much longer reaction time was required for achieving good yields in the oxidation of substituted benzyl-amines (Table 3, entries 2–4). Surprisingly, 2-thiophenemethyl-amine gave 99.3% conversion and 97.0% selectivity in 3 h (Table 3, entry 5). Aliphatic amines showed very low conversion (Table 3, entries 6–7), as was due to the lack of active groups attached to NH2 of the aliphatic amines [27]. According to the evaluation results of the catalysts, benzyl-amine could produce product imine, as well as by-product benzaldehyde, benzonitrile, benzamide during the reaction. When benzylamine was highly or completely converted, the amount of benzonitrile would quickly increase. According to the literature [27], benzylamine would form the imine intermediate during the oxidation process, but it was not detected in the reaction. It might be because the imine intermediate from the oxidation of benzyl-amine was very unstable, and it was hydrolyzed to form benzaldehyde by the water formed in situ. According to literature [27] and experiments, we first proposed the more possible reaction pathway followed the oxidation of amines as shown in Scheme 1. In general, the oxidative dehydrogenation pathway was suggested for the oxidation of amines. An imine intermediate was formed from initial amine (step Ⅰ), and then hydrolyzed by water generated in situ to give an aldehyde (step Ⅱ). The formed aldehyde instantly reacted with available amine to yield the final imine (step Ⅲ). Simultaneously, the by-product benzonitrile was formed by further oxidation of a very small amount of imine intermediate (step Ⅵ). The trace benzonitrile could be hydrolyzed to give the amide (step Ⅶ). When the initial amine molecules were almost converted, the aldehyde formed from the reverse reaction of imine could not be effectively condensed with the trace amine, but could be condensed with ammonia molecules generated in situ to form imine intermediates (step V). Reversible amines were also oxidized to imine intermediates (step Ⅰ). The small part of imine intermediates were hydrolyzed to form aldehydes (step Ⅱ), and the other part were further oxidized to form nitriles (step Ⅵ), and a small amount of nitriles were hydrolyzed to form amides (step Ⅶ). As the process was continuously circulated, the selectivity of imine continued to decrease, and the selectivity of benzonitrile and benzamide continues to increase. The speculation was well confirmed by the experimental results.
|
|
Table 3 The synthesis of imines from various amines by the M-4 catalyst.a |

{kind=link}
{kind=link}
{kind=link}

|
Download:
|
| Scheme 1. Possible reaction pathway for the oxidation of amines. | |
{kind=link}
It was well known that the catalytic activity of manganese oxide was determined by several factors such as lattice oxygen, adsorbed oxygen, oxygen vacancy, surface oxidation state of manganese, mobility of lattice oxygen, texture properties, and so on [1, 19, 27-29, 32, 33]. By comparing the structure-activity relationship, we found that Mn4+ in manganese oxide might be strongest active sites, but easily led to excessive oxidation of benzylamine to benzonitrile. Mn3+ might be the main active sites of benzylamine oxidation to imine, which was also confirmed by the literature [19]. It was surprisingly found that the selectivity of imine was better when the Mn3+/Mn4+ ratio was higher (Tables 1 and 2, Fig. S9 in Supporting information). Mn2+ in manganese oxalate almost showed no catalytic activity (Table 2, entry 3). However, higher content of Mn2+ in manganese oxide could generate more oxygen vacancies, and promoted aerobic oxidation [27]. M-3 with highest specific surface area (304.7 m2/g) showed highest Mn4+ content (0.386) compared with M-4 (0.254) and M-5 (0.273) (Table S1 and Table 1), resulting in highest catalytic activity and easy oxidizing imine intermediates to nitrile (Table 2, Fig. 3). Although it gave lower Mn4+ content (0.254), M-4 with sharply reduced surface area (28.4 m2/g) showed slight reduction in catalytic activity. This was because Mn2+ content (0.260) significantly increased to generate more oxygen vacancies, which promoted aerobic oxidation. Higher water molecules (0.143) among surface oxygen species on the M-4 surface promoted hydrolysis of the imine intermediate to aldehyde (Scheme 1, step Ⅱ), which further reacted with another amine to form the product imine (Scheme 1) [19]. This might be another important reason for higher activity and selectivity of M-4. The addition of trace water increased the selectivity of the imine in the process of using M-3 as the catalyst (Table 2, entries 4 and 7), which further confirmed that water played a positive role in the reaction process. Trace water weakened further oxidation of the imine intermediate to form a nitrile (Scheme 1, step Ⅵ). M-5 gave the lowest catalytic activity, which might be related to lower Mn4+ content (0.273), lowest surface area (19.4 m2/g), lowest content of water molecules (0.045) among surface oxygen species. Manganese oxides displayed a two-step reduction (Mn2O3 (MnO2) → Mn3O4 → MnO) (Fig. S4 from H2-TPR in Supporting information) [34]. The lattice oxygen mobility of the catalysts could be associated with the ease of the manganese oxides reducibility, which played an important role on the catalytic oxidation reactions [16, 29, 34]. Fig. S4 implied that M-5 possessed lowest lattice oxygen mobility, which might also be the important reasons for lowest activity of M-5. In addition, the selectivity of imines might be related to pore diameter (Table S1, Fig. S1, Table 2), and larger pore size facilitated the diffusion of larger imine molecules.
According to experimental results and the literature [19], We suggested a mechanism for the oxidative self-coupling of amines catalyzed by M-4 (Scheme S1 in Supporting information). An electron of adsorbed amine molecules was transferred to Mn4+/Mn3+ (Mn4+/Mn3+ → Mn3+/Mn2+), and an α-C–H proton and N H proton were eliminated to generate the RCH = NH intermediate (Scheme S2 in Supporting information). In this process, the rate-determining step was the abstraction of α-C–H proton (Scheme S2). One-electron reduction of Mn4+/Mn3+ could facilitate release of lattice oxygen. The reduced Mn species could be re-oxidized to Mn4+/Mn3+ by O2 decomposed from H2O2 in the aid of manganese oxide [35]. The aerobic atmosphere was crucial for the catalytic activity because replenishment of the consumed lattice oxygen required aerial oxygen. This was consistent with the observation of lower catalytic efficiency under nitrogen atmosphere (Table 2, entry 9).
In summary, we had successfully developed the manganese oxide catalyst showing higher selectivity of amines to imines by simple oxalate route. Characterization of M-4 showed that it had micro-rod morphology, large average pore diameter, moderate lattice oxygen mobility, highest content of water in surface oxygen species, high Mn3+/Mn4+ ratio, and was mainly composed of cubic crystal Mn2O3. The enhanced selectivity of M-4 catalyst was attributed to the high Mn3+/Mn4+ ratio and content of water among surface oxygen species. Besides, various substituted imines could also be synthesized with improved selectivity using the synthetic method introduced herein. Further investigations were underway.
AcknowledgmentsFinancial support from the National Natural Science Foundation of China (NSFC, Nos. 21776258, 21476207, 91534113, 21406199, 21566013, 21875220), Program from Science and Technology Department of Zhejiang Province (Nos. 2015C31042, LY17B060006), Education Science Planning Project of Jiangxi Province (No. 18YB243), and Natural Science Foundation of Jiangxi Province (No. 20181BAB216032) are gratefully acknowledged.
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.09.007.
| [1] |
B. Chen, L. Wang, S. Gao, ACS Catal. 5 (2015) 5851-5876. DOI:10.1021/acscatal.5b01479 |
| [2] |
B. Dutta, S. March, L. Achola, et al., Green Chem. 20 (2018) 3180-3185. DOI:10.1039/C8GC00862K |
| [3] |
M.T. Schümperli, C. Hammond, I. Hermans, ACS Catal. 2 (2012) 1108-1117. DOI:10.1021/cs300212q |
| [4] |
H. Naeimi, F. Salimi, K. Rabiei, J. Mol. Catal. A Chem. 260 (2006) 100-104. DOI:10.1016/j.molcata.2006.06.055 |
| [5] |
K.C. Nicolaou, C.J.N. Mathison, T. Montagnon, Angew. Chem. Int. Ed. 42 (2003) 4077-4082. DOI:10.1002/anie.200352076 |
| [6] |
W. He, L. Wang, C. Sun, et al., Chem. -Eur. J. 17 (2011) 13308-13317. DOI:10.1002/chem.201101725 |
| [7] |
L. Zhang, W. Wang, A. Wang, et al., Green Chem. 15 (2013) 2680-2684. DOI:10.1039/c3gc41117f |
| [8] |
G. Yuan, M. Gopiraman, H.J. Cha, et al., J. Ind. Eng. Chem. 46 (2017) 279-288. DOI:10.1016/j.jiec.2016.10.040 |
| [9] |
B. Zhu, M. Lazar, B.G. Trewyn, et al., J. Catal. 260 (2008) 1-6. DOI:10.1016/j.jcat.2008.08.012 |
| [10] |
G. Chu, C. Li, Org. Biomol. Chem. 8 (2010) 4716-4719. DOI:10.1039/c0ob00043d |
| [11] |
L. Al-Hmoud, C.W. Jones, J. Catal. 301 (2013) 116-124. DOI:10.1016/j.jcat.2013.01.027 |
| [12] |
A. Santiago-Portillo, J.F. Blandez, S. Navalon, et al., Catal. Sci. Technol. 7 (2017) 1351-1362. DOI:10.1039/C6CY02577C |
| [13] |
H. Huang, J. Huang, Y.M. Liu, et al., Green Chem. 14 (2012) 930-934. DOI:10.1039/c2gc16681j |
| [14] |
C. Xu, H. Liu, D. Li, et al., Chem. Sci. 9 (2018) 3152-3158. DOI:10.1039/C7SC05296K |
| [15] |
Y. Xu, Y. Chen, W.-F. Fu, Appl. Catal. B:Environ. 236 (2018) 176-183. DOI:10.1016/j.apcatb.2018.03.098 |
| [16] |
S. Biswas, A.S. Poyraz, Y. Meng, et al., Appl. Catal. B 165 (2015) 731-741. DOI:10.1016/j.apcatb.2014.10.055 |
| [17] |
B. Dutta, V. Sharma, N. Sassu, et al., Green Chem. 19 (2017) 5350-5355. DOI:10.1039/C7GC01919J |
| [18] |
D. Biswanath, B. Sourav, S. Vinit, et al., Angew. Chem. Int. Ed. 128 (2016) 2211-2215. DOI:10.1002/ange.201508223 |
| [19] |
S. Biswas, B. Dutta, K. Mullick, et al., ACS Catal. 5 (2015) 4394-4403. DOI:10.1021/acscatal.5b00325 |
| [20] |
K. Mullick, S. Biswas, C. Kim, et al., Inorg. Chem. 56 (2017) 10290-10297. DOI:10.1021/acs.inorgchem.7b01177 |
| [21] |
K. Mullick, S. Biswas, A.M. Angeles-Boza, et al., Chem. Commun. 53 (2017) 2256-2259. DOI:10.1039/C6CC09095H |
| [22] |
S. Biswas, K. Mullick, S.Y. Chen, et al., ACS Catal. 6 (2016) 5069-5080. DOI:10.1021/acscatal.6b00717 |
| [23] |
P. Venkataswamy, K.N. Rao, D. Jampaiah, et al., Appl. Catal. B:Environ. 162 (2015) 122-132. DOI:10.1016/j.apcatb.2014.06.038 |
| [24] |
C.H. Kuo, I.M. Mosa, A.S. Poyraz, et al., ACS Catal. 5 (2015) 1693-1699. DOI:10.1021/cs501739e |
| [25] |
S.L. Suib, J. Mater. Chem. 18 (2008) 1623-1631. DOI:10.1039/b714966m |
| [26] |
Z. Zhang, F. Wang, M. Wang, et al., Green Chem. 16 (2014) 2523-2527. DOI:10.1039/C3GC42312C |
| [27] |
P. Sudarsanam, B. Hillary, M.H. Amin, et al., Appl. Catal. B:Environ. 185 (2016) 213-224. DOI:10.1016/j.apcatb.2015.12.026 |
| [28] |
F. Chen, S. Zhao, T. Yang, et al., Acta Phys. Chim. Sin. 35 (2019) 775-786. |
| [29] |
W. Tang, X. Wu, D. Li, et al., J. Mater. Chem. A 2 (2014) 2544-2554. DOI:10.1039/C3TA13847J |
| [30] |
L.V. Stebounova, N.I. Gonzalez-Pech, T.M. Peters, et al., Environ. Sci. Nano 5 (2018) 696-707. DOI:10.1039/C7EN01046J |
| [31] |
Y. Wu, R. Feng, C. Song, et al., Catal. Today 281 (2017) 500-506. DOI:10.1016/j.cattod.2016.05.024 |
| [32] |
X.B. Huang, L.P. Liu, H.Y. Gao, et al., Green Chem. 19 (2017) 769-777. DOI:10.1039/C6GC02065H |
| [33] |
G. Liu, L. Sun, J. Liu, et al., Mol. Catal. 440 (2017) 148-157. DOI:10.1016/j.mcat.2017.07.017 |
| [34] |
A.S. Poyraz, W. Song, D. Kriz, et al., ACS Appl. Mater. Interface 6 (2014) 10986-10991. DOI:10.1021/am502846e |
| [35] |
V.D. Makwana, Y.C. Son, A.R. Howell, et al., J. Catal. 210 (2002) 46-52. DOI:10.1006/jcat.2002.3680 |