 2019, Vol. 30
2019, Vol. 30 


b State Key Laboratory of Chemical Resource Engineering, Beijing University of Chemical Technology, Beijing 100029, China;
c Beijing Key Laboratory of Energy Conversion and Storage Materials, College of Chemistry, Beijing Normal University, Beijing 100875, China
Burning of fossil fuels provides about 85% of the world's energy supply while emitting large amounts of CO2 and other hazardous into atmosphere causing "greenhouse" effect and air pollution. Thus, technological innovation on carbon capture, utilization and storage (CCUS) is one of the most important strategy to encounter the global climate change and has attracted widespread attentions. Electrochemical CO2 reduction reaction (CO2RR) can convert CO2 into value-added chemicals and fuels, which can be used as industrial feedstocks providing a "clean" and promising way to lower the global carbon emission [1-3]. Therefore, CO2RR driven renewable energy resources such as wind energy, tidal energy and solar energy have been regarded as a key technology for mitigating climate problems and finally achieve carbon neutral circulation. The undertaking of CO2RR basically includes the four steps: (ⅰ) CO2 adsorption on dynamic destinations, (ⅱ) formation of intermediates, such as CO2- or HCO2- by the actuation of carbon monoxide (CO), (ⅲ) breaking of C-O bond involving the H+ and e- exchange steps, and (ⅳ) desorption of reduction species from the dynamic locations [4-6]. Therefore, the noteworthy vitality hindrances amid the adsorption and activation steps assume a pivotal job for CO2RR and require a quantitative appraisal on the electrocatalysts which can productively adsorb, activation and convert CO2 into valuable chemicals. To efficiently converting CO2 to target products, a large number of heterogeneous catalysts have been developed to propel the electrochemical process with admirable activity and selectivity [7, 8]. Despite these efforts, however, there are still many limitations to perform the large-scale CO2RR application. For example, drawbacks exist in large over potentials, low current densities, and poor Faraday current efficiencies during electrochemical reduction due to higher energy barriers for CO2 adsorption and activation. Therefore, it is particularly urgent to design a high-efficiency CO2 reduction catalyst having the ability to adsorb and activate CO2 molecules on the surface.
Recently, O-vacancies of metallic oxide have been found to offer the rich e- charge appropriation and critical surface adsorption capacity to facilitate the CO2 adsorption and activation [9-11]. Further, the O-vacancies in metal oxides advances the pi-back attaching to the adsorbates that influence the d-band energy levels of electrocatalyst adjusting the coordination condition of dynamic site and advance the arrangement of activated COOH* response transitional [9, 10]. However, the profound understanding of O-vacancies for CO2RR in atomic scale is still at the infancy, which is important for the catalyst optimization. Some specialists are focusing to reveal the science between O-vacancies and CO2RR catalysis by setting up some zinc oxides (ZnOx) and cobalt oxides (CoxOy) and suggested that the atomic thickness of these oxides support the thermal stability of the structure as well as empowers most of O-vacancies appropriation on the outside of the catalysts that helps the electron-contributor dynamic destinations to catch the CO2 molecules and activate the stable C = O moiety [9, 12]. Tin oxide (SnOx) materials have been confirmed to have high activity for CO2RR [13, 14]. In SnO2, Sn atoms with outer layer electron configuration of 5s25p2 donating 4 electrons to 2 oxygen atoms with electronic configuration of 2s22p4 forming chemical bonds, which can form O-vacancies in crystal structure by various means of high temperature reduction (H2, Mg, Al), plasma and laser treatment [15]. It offers a great opportunity to study the electronic effect of metal oxides on CO2RR synergist execution by changing their O-vacancies concentration. In any case, rare study reports on the annealing temperature reliance of O-vacancies concentration in SnO2 and accounting which for CO2RR performance to comprehend the electrochemical activity and selectivity with chemical composition and structure of this material.
Herein, we quantitatively investigated the influence of O-vacancies on the CO2RR by comparing a series of nanoporous tin oxide (SnOx) with varied degree of O-vacancies via thermal treatment. The annealing temperature played important role in balancing the desorption of oxygen species and the continuous oxidation of SnOx, and thereby finely controlling the O-vacancies. The as-prepared nanoporous SnOx with 300 ℃ treatment was found to be highest O-vacant material and showed an impressive CO2RR activity and selectivity towards the conversion of CO2 into formic acid up to 88.6%. The ideal performance of the O-vacancies rich SnOx-300 material can be ascribed to the high delocalized electron density inducing much enhanced adsorption of CO2 and benefiting the reduction with high selectively forming of formic acid. The strategy of O-vacancies manipulation would provide deeper insights into rational optimization of electrocatalyst for CO2RR.
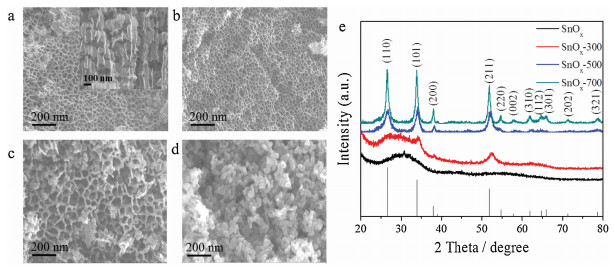
The synthesis of SnOx materials with different concentrations of O-vacancies were accomplished by means of heat treatment at different temperature as represented in Scheme 1. Clean tin foil was firstly electro-oxidized to form SnOx, and then annealed via 30 min thermal treatment at different temperatures of 300 ℃, 500 ℃, and 700 ℃, respectively, labled as SnOx-300, SnOx-500, and SnOx-700. As revealed by SEM image (Fig. 1a), the surface of tin foil becomes honeycomb like nanoporous structure with the pore size of 30-60 nm after the electrooxidation at a high current density of 3 A/cm2, which can be ascribed to the induction of rapid evolution of oxygen bubble accompanied with the formation of tin oxide during the anodic oxidation process [16]. The surface oxidation layer is found to be amorphous with no obvious diffraction patterns (Fig. 1e). The porous structure would create more active sites and benefit the transportation of gas and electrolyte, offering enough conductivity to the electrocatalytic dynamic SnOx materials for the electrochemical response processes [17, 18]. After the thermal treatment, the surface morphology and pore size of SnOx-300 remains essentially unchanged (Fig. 1b), but the crystalinity increases according to the appearance of weak diffraction patterns at 26.61, 33.89, 37.95 and 51.78 degrees indexed into typical (110), (101), (200) and (211) peaks (PDF# 77-0447). As the thermal treament temperature rise, the nanochannels gradually break down and reconstruct as indicated by the enlarged pore size for SnOx-500 with small amount of particles (Fig. 1c) and totally nanoparticles with the size of 30-50 nm for SnOx-700 (Fig. 1d), the diffractionpeak intensity significantly increases [19, 20]. In order to further clarify the changes of the porosity, the Brunauer-Emmett-Teller (BET) analysis were carried out and the results are shown in Fig. S1 and Table S1 (Supporting information). Hysteresis was observed in all the SnOxmaterials, which is attributedtothe capillarycondensation in the mesoporous materials. The pore analysis demonstrated that the tendency of pore diameter decreases with processing temperature elevation, which is consistent with the SEM observation. Furthermore, the SnOx-300 displayed the highest specific surface area compared with other three samples, which indicated that SnOx-300 can support more active site distribution.

|
Download:
|
| Scheme 1. Schematic illustration of the synthesis of as-prepared SnOx materials. | |
{kind=link}

|
Download:
|
| Fig. 1. SEM images of (a) SnOx, (b) SnOx-300, (c) SnOx-500 and (d) SnOx-700. (e) XRD patterns of all SnOx samples. | |
{kind=link}
To confirm the influence of thermal treatment on O-vacancies, we performed X-ray photoelectron spectroscopy (XPS) analysis, which was shown in Figs. 2a–d and Fig. S2 (Supporting information). The O 1s spectra can be divided into three components at 530.6, 531.7 and 532.6 eV, which attributing to different oxygen species of Sn-O bond in crystal structure, O-vacancies, and hydroxyl of the surface adsorbed moisture, respectively [14, 21]. It can be clearly seen that the XPS peak intensity of O-vacancies at 531.7 eV is higher for SnOx-300 compared to pristine SnOx after heating at 300 ℃ and further decreases with the rising of temperature.

|
Download:
|
| Fig. 2. The O 1 s XPS spectra of (a) SnOx, (b) SnOx-300, (c) SnOx-500 and (d) SnOx-700. (e) The calculated ratio of integral areas for oxygen vacancies from O 1s XPS spectra for all the samples and (f) Sn 3d XPS spectra. | |
{kind=link}
The concentration variation of O-vacancies can also be verified by the histogram of integral area ratios (Fig. 2e). Thus, SnOx-300 appeared to have the highest O-vacancy amount of 37%. The O-vacancies are normally with positive valence and can be utilized as regular electron acceptors [22, 23]. Accordingly, under the electron pulling effect of surface O-vacancies, there is obvious binding energy shift to higher value for Sn 3d in SnOx-300, which is in tune with the O 1s XPS analysis further indicating the highest O-vacancy concentration.
Given the quantitatively verification of O-vacancies, the linear sweep voltammetry (LSV) curves performed in CO2 saturated 0.5 mol/L KHCO3 electrolyte (Fig. 3a). As expected, SnOx-300 exhibited higher CO2RR current density and onset potential than other opponents. The trend affirms the O-vacancies association with CO2RR by mean of proportionality. The profitability and selectivity were checked by the gas chromatography (GC) and nuclear magnetic resonance (NMR) measurements. CO and H2 were found to be the gas products in all instances of SnOx materials, and the liquid product was formic acid (Fig. 3b). The sum of faradaic efficiencies of these three products were already close to 100% (Fig. S3 in Supporting information). For SnOx-300 materials, the faradic efficiency (FE) can reach up to 88.6% at the potential of -0.98 V, much higher than the other three samples of 76.5%, 57.4% and 45.4%, respectively. In order to understand the correlation between catalytic activity and the type of the oxygen species, the trend in maximum FE for CO along with its corresponding potential versus concentrations of different oxygen species in four SnOx catalysts is investigated (Fig. 3c). The maximum FE for HCOOH increases and the corresponding potential shifts anodically as the oxygen vacancies content increases, whereas no such a trend is observed for Sn-O and adsorbed O. These results suggest that oxygen vacancy is the most active site for CO2 reduction. Fig. 3d demonstrates the fractional current density curves of formic acid for the four samples, and SnOx-300 is also the highest one and the trend is in accord with the oxygen vacancy concentration. Moreover, the ratio of FECO2 and FEH2 over the four electrocatalysts display a similar trend with the above results (Fig. S4 in Supporting information). The reaction kinetics of the four samples was demonstrated by their corresponding Tafel slopes (Fig. 3e). As well known, the adsorption of CO2 molecules is with the highest energy barrier and the keya to kinetic control [9, 10]. O-vacancies help capture the CO2 anion radical (CO2*-) and hence favor the subsequent hydrogeneration process [11], which coincides the lowest Tafel slope of 115.21 mV/dec for SnOx-300 while that of SnOx, SnOx-500 and SnOx-700 were found to be 143.69 mV/dec, 230.09 mV/dec and 308.29 mV/dec, respectively.

|
Download:
|
| Fig. 3. Electrochemical CO2 reduction activities of SnOx, SnOx-300, SnOx-500, SnOx-700. (a) LSV curves obtained using four SnOx catalysts in CO2-saturated aqueous 0.5 mol/L KHCO3 electrolyte solutions with a 20 mV/s scan rate. (b) FEs of HCOOH at each given potential for 1 h. (c) The change curves of onset potential and the maximum FE of HCOOH with relative proportion of oxygen species. (d) Partial current densities for the production of HCOOH. (e) Tafel slopes. | |
{kind=link}
In addition, considering the structural variation is from intact 3D porous structure for SnOx-300 to the stacked granular structure for SnOx-700, the active sites and conductivity would also be affected. As evidenced by the electrochemical impedance spectra recording in 0.5 mol/L KHCO3 electrolyte at -0.98 V (Fig. S5 and Table S2 in Supporting information), the surface charge resistance of SnOx, SnOx-300, SnOx-500 and SnOx-700 were 5.898 ohm, 4.06 ohm, 7.654 ohm and 9.634 ohm, respectively, indicating the ideal conductivity of SnOx-300. Further, because of the total collapse of the pore structure for SnOx-700, there would be a huge decline for stacked granular structure to contact with the electrolyte, inducing a relative high increasement in the contact resistance. Similarly, the electrochemical surface areas can be utilized to describe the quantity of active sites and they can be approximated by utilizing electric capacitance computations [24]. Fig. S6 (Supporting information) demonstrates the CV curves and the calculation of electric double layer capacitance at various tin oxides in Ar-saturated 0.5 mol/L KHCO3 electrolyte. The outcomes demonstrated that SnOx-300 appeared to have the largest electric double layer capacitance agree with the specific surface area results, showing great accessibility of active sites for CO2RR. Hence, the intact porous crystalline structure characteristic of SnOx-300 endows ideal conductivity and abundant active sites exposure, contributing the good performance for CO2 electrochemical conversion, as well as O-vacancies [25]. The stability test was evaluated in a CO2-saturated 0.5 mol/L KHCO3 solution using a potential of -0.98 V (Fig. S7 in Supporting information). The SnOx-300 electrode showed a good stability, the FEHCOOH remained 80% after electrolysis for 18 h.
Fig. 4 demonstrates the proposed electrochemical transformation of CO2 to formic acid including adsorption, activation and desorption steps. Under the variable reaction conditions, CO2 was first adsorbed on the surface of the catalysts and converted into formic acid by the accompanying e-exchange steps where the initial step is the rate determine step and includes two types of electrochemical absorption as O-binding (CO2*-) or C binding (*COO-) of CO2 with the catalysts surface through charge transfer. (ⅰ) The adsorbed CO2*- by O binding on active sites can accelerate the formation of adsorbed HCOO* species, which was a crucial intermediate for CO2RR to HCOOH after binding with proton and further electron transfer [26]. (ⅱ) The C binding *COO- further includes proton to form a *COOH intermediates which further accept protons and electrons. This consequent progress can continue with two paths: (a) hydrogenation path, which includes adsorption of H on the surface of catalyst and consolidate with Catom to form formic acid; and (b) drying out path, this path includes the nucleophilic attack of the —OH with proton, bond break from C atom and desorption of CO [27].

|
Download:
|
| Fig. 4. Possible reaction pathways for the electroreduction of CO2 to produce HCOOH and CO using Sn based catalysts. | |
{kind=link}
Likewise, as shown in Fig. S3, the Faraday efficiency bend for the formation of CO by CO2RR for all tin oxide catalysts. It tends to be seen from the assumption that SnOx, SnOx-500 and SnOx-700 electrode catalysts with various concentration of O-vacancies showed lower CO development action, while the SnOx-300 catalyst with the most elevated O-vacancies fixation has a CO arrangement efficiency of under 1%, diminishing the dimension of hydrogen development essentially. Joined with the test information of electrochemical portrayal of the system for the conversion of CO2 into formic acid, it can be affirmed that the nearness of O-vacancies can fundamentally expand the appropriation of CO2*- radicals anion framed on the outside of the catalyst surface by O adsorption after the electrons are adsorbed. The proportion and consequent proton movements are controlled by hydrogenation to produce formic acid. Also, Xie et al. [9] utilized DFT hypothesis to reenact the difference in free vitality of CO2 arrangement and interaction with the O-vacancies sites. While affirming the above perspectives, they likewise demonstrate that the adsorption and activation of CO2 on O-vacancies also affects the stability of the reaction intermediates, which breaks the scale relationship between the intermediates in CO2RR process. In our case, O-vacancies in tin oxides not only essentially promote the CO2*- radical formation in the O-binding mode but also upgrade the settling impact of the catalysts on HCOO* transitional, thus contributing to the high formate selectivity of SnOx-300 in CO2RR.
In conclusion, the nanoporous tin oxide electrode materials were set up by electrooxidation, and the convergence of O-vacancy in tin oxides was constrained by the thermal treatment in air at various temperatures. Further, leading a progression of electrochemical tests, it is affirmed that the centralization of O-vacancies in the catalyst surface has an immediate relationship with the CO2RR movement and selectivity. The presence of O-vacancies cannot just build the extent of CO*- radical anion yet additionally breaks the inalienable size relationship of intermediates in CO2 reduction by settling HCOO* intermediates. The CO2RR action of SnOx-500 and SnOx-700 materials were thought about. Since, these two materials appeared to have changed auxiliary morphology and comparable grouping of O-vacancies, the watched variety in CO2RR action can be credited to the morphology impact. Consequently, the O-vacancies rich and honeycomb nanoporous organized SnOx-300 material catalyzed the reduction of CO2 into formic acid at -0.98 V, appearing huge Faraday efficiency (88.6%) and partial current density (15 mA/cm2). Through the above research, a controlled concentration of O-vacancies with normal morphology has been acknowledged by basic techniques, which can be filled in as a source of perspective for the further plan and scale-up of electrocatalysts for the productive catalysis of CO2 to formic acid later on.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China, National Key Research and Development Project (No. 2016YFF0204402), the Program for Changjiang Scholars and Innovative Research Team in the University, and the Fundamental Research Funds for the Central Universities, and the longterm subsidy mechanism from the Ministry of Finance and the Ministry of Education of PRC.
Appendix A. Supplementary dataSupplementary material related to this article can be found, inthe online version, at doi:https://doi.org/10.1016/j.cclet.2019.07.028.
| [1] |
W. Zhu, R. Michalsky, Ö. Metin, et al., J. Am. Chem. Soc. 135 (2013) 16833-16836. DOI:10.1021/ja409445p |
| [2] |
S. Zhang, P. Kang, T.J. Meyer, J. Am. Chem. Soc. 136 (2014) 1734-1737. DOI:10.1021/ja4113885 |
| [3] |
X. Huang, T. Cao, M. Liu, G. Zhao, J. Phys. Chem. C 117 (2013) 26432-26440. DOI:10.1021/jp408630s |
| [4] |
H. Mistry, R. Reske, Z. Zeng, et al., J. Am. Chem. Soc. 136 (2014) 16473-16476. DOI:10.1021/ja508879j |
| [5] |
W. Zhu, Y. Zhang, H. Zhang, et al., J. Am. Chem. Soc. 136 (2014) 16132-16135. DOI:10.1021/ja5095099 |
| [6] |
S. Ma, M. Sadakiyo, M. Heima, et al., J. Am. Chem. Soc. 139 (2017) 47-50. DOI:10.1021/jacs.6b10740 |
| [7] |
A. Vasileff, C. Xu, Y. Jiao, Y. Zheng, S. Qiao, Chemistry 4 (2018) 1809-1831. DOI:10.1016/j.chempr.2018.05.001 |
| [8] |
S. Lee, J. Lee, ChemSusChem 9 (2016) 333-344. DOI:10.1002/cssc.201501112 |
| [9] |
S. Gao, X. Jiao, Z. Sun, et al., Angew. Chem. Int. Ed. 55 (2016) 698-702. DOI:10.1002/anie.201509800 |
| [10] |
J. Zhang, R. Yin, Q. Shao, T. Zhu, X. Huang, Angew. Chem. Int. Ed. 58 (2019) 5609-5613. DOI:10.1002/anie.201900167 |
| [11] |
Z. Geng, X. Kong, W. Chen, et al., Angew. Chem. Int. Ed. 57 (2018) 6054-6059. DOI:10.1002/anie.201711255 |
| [12] |
S. Gao, Z. Sun, W. Liu, et al., Nat. Commun. 8 (2017) 14503-14511. DOI:10.1038/ncomms14503 |
| [13] |
F. Li, L. Chen, G.P. Knowles, D.R. MacFarlane, J. Zhang, Angew. Chem. Int. Ed. 55 (2016) 1-6. DOI:10.1002/anie.201510990 |
| [14] |
R. Dai, X. Lu, W.H. Saputera, Y.H. Ng, R. Amal, ACS Sustain. Chem. Eng. 6 (2018) 1670-1679. DOI:10.1021/acssuschemeng.7b02913 |
| [15] |
M.V. Pirovano, A. Hofmann, J. Sauer, Surf. Sci. Rep. 62 (2007) 219-270. DOI:10.1016/j.surfrep.2007.03.002 |
| [16] |
H. Shin, J. Dong, M. Liu, Adv. Mater. 16 (2004) 237-240. DOI:10.1002/adma.200305660 |
| [17] |
L. Fan, Z. Xia, M. Xu, Y. Lu, Z. Li, Adv. Funct. Mater. 28 (2018) 1706289-1706296. DOI:10.1002/adfm.201706289 |
| [18] |
S. Chappel, A. Zaban, Sol. Energy Mater. Sol. Cells 71 (2002) 141-152. DOI:10.1016/S0927-0248(01)00050-2 |
| [19] |
G.E. Unni, S. Sasi, A.S. Nair, J. Energy Chem. 25 (2016) 481-488. DOI:10.1016/j.jechem.2016.02.008 |
| [20] |
C. Haw, W. Chiu, N.H. Khanis, et al., J. Energ. Chem. 25 (2016) 691-701. DOI:10.1016/j.jechem.2016.04.006 |
| [21] |
B. Zhang, L. Wang, Y. Zhang, Y. Ding, Y. Bi, Angew. Chem. Int. Ed. 57 (2018) 2248-2252. DOI:10.1002/anie.201712499 |
| [22] |
Y. Yang, Y. Wang, S. Yin, Appl. Surf. Sci. 420 (2017) 399-406. DOI:10.1016/j.apsusc.2017.05.176 |
| [23] |
J.J. Yang, M.D. Pickett, X.M. Li, et al., Nat. Nanotechnol. 3 (2008) 429-433. DOI:10.1038/nnano.2008.160 |
| [24] |
Y. Duan, F. Meng, K. Liu, et al., Adv. Mater. 30 (2018) 1706194-1706200. DOI:10.1002/adma.201706194 |
| [25] |
L. Wang, W. Zhang, X. Zheng, et al., Nat. Energy 2 (2017) 869-876. DOI:10.1038/s41560-017-0015-x |
| [26] |
A.A. Peterson, J.K. Nørskov, J. Phys. Chem. Lett. 3 (2012) 251-258. DOI:10.1021/jz201461p |
| [27] |
J. Schneider, H. Jia, J.T. Muckerman, E. Fujit, Chem. Soc. Rev. 41 (2012) 2036-2051. DOI:10.1039/C1CS15278E |