 2019, Vol. 30
2019, Vol. 30 


b College of Materials Science and Engineering, Fuzhou University, Fuzhou 350116, China;
c School of Resources and Chemical Engineering, Sanming Institute of Fluorochemical Industry, Sanming University, Sanming 365004, China
With the rapid development of green energy technologies, eco-friendly and renewable energy storage devices plays an extremely important role in our daily life, including lithium-ion batteries [1], super-capacitors [2] and solar cells [3]. Organic lithium-ion batteries (OLIBs) are considered as one of promising candidates for the next generation electronical energy storage [4]. Organic compounds as the redox-active materials are mainly attributed to the advantage of their tunable structure and properties, sustainability and low cost [5]. Among various organic electrodes redox-active materials, carbonyl-containing organic molecules have been used widely in organic batteries owing to their outstanding electrochemical performances [6]. Aromatic diimides are an emerging class of carbonyl-containing redoxactive species for lithium-ion batteries, and their battery performances could be well tuned by molecular engineering [7]. For example, the charge/discharge rates of these unique redox-active organic molecules (ROMs) could be enhanced by their electronic conductivity, which has been easily achieved by the extension of π-systems [8]. Accordingly, in the past years, our groups have extended the well-known perylene diimide (PDI) ROMs at the lateral positions to obtained a series of benzo[ghi] perylenetriimides with different structures and indicated that such lateral π-extension has indeed increased their battery performances in comparison with those of PDIs, including charge/discharge rates, conductivities and cycling lives [9]. However, it has simultaneously led to a hypsochromic shift of absorption bands, which implied that the energy gaps (Eg) of their HOMO-LUMO levels were raised and thus lowered their capacity utilizations.
In contrast, the π-extension of aromatic diimides along the molecular long axis (vertical direction) has been demonstrated to be an efficient strategy to achieve a bathochromic shift of the absorption maxima, for example, ca. 100 nm bathochromic shifts per naphthalene unit [10]. Semi-empirical calculations have revealed that in rylene diimides the LUMO level is affected only slightly, whereas the HOMO level rises [11]. As a result of such vertical π-extensions, the capacity utilizations of aromatic diimides are supposed to be higher but the fluctuations of their redox potentials to be limited. More importantly, due to the different Clar's structures [12] possessed in their reduction states (one-electron as well as two-electron reductions), the chemical stabilities of their reduction intermediates should also be different. To validate these deductions, herein we have selected three vertically π-extended aromatic diimides, naphthalene diimide (NDI), PDI and terrylene diimide (TDI), as shown in Scheme 1. When served as electrode materials in organic lithium ion batteries, the electrochemical studies indicated that their cycling stabilities are strongly dependent on the sizes of their π-systems [13]. As expected, three homologues exhibited three totally different charge/discharge profile, specifically, two-step equivalent well-resolved charge/discharge profiles for NDIs, but only one-step charge/discharge profile for PDIs, and one-step distorted charge/discharge profile for TDIs. This difference in electronic redox mechanisms has also been further rationalized by the chemical stabilities of their reduction states through density functional theory (DFT) calculations.

|
Download:
|
| Scheme 1. Chemical structures of NDI, PDI and TDI. | |
{kind=link}
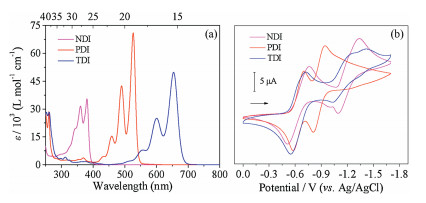
As stated above, all the organic active electrode materials (NDI, PDI and TDI), were prepared according to previous reports [14]. The UV-vis absorption spectra of NDI, PDI and TDI were investigated in CH2Cl2 at room-temperature, which exhibited the classical absorption features with two intense absorption bands from 400 nm to 700 nm as well as two less distinct shoulder bands at the short wavelength sides, assigned to the energy transitions S0 to S1 vibrational states [15]. Specifically, NDI shows absorption maxima at 380 nm (35426 L mol-1 cm–1) while the PDI and TDI exhibits the bathochromic and absorption maxima at 527 nm (70698 L mol-1 cm–1) and 654 nm (49610 L mol-1 cm–1), respectively. Accordingly, the optical energy-band gaps calculated from the onsets of absorption spectra for the NDI, PDI and TDI were 3.26 eV, 2.35 eV and 1.90 eV, respectively. In addition, according to the reported spectro-electrochemical results by Saha's and Hasobe's groups [16], all three diimides could be further reduced to form their stable radical anions/ dianions, which have motivated to explore their electrochemical applications.
The electrochemical properties of three aromatic diimide homologues have been investigated by cyclic voltammograms (CVs) in DCM. As shown in Fig. 1b, two well-defined single-electron reversible reduction waves have been observed in the solvents for the all three diimides NDI, PDI and TDI. For NDI, the two half-wave reduction potentials (E1/2)were estimatedtobe-0.64 V and-1.22 V relative to Ag/AgCl couple, which are resulted from the formation of a charge delocalized radical anion [NDI]·- and dianion [NDI]2-, respectively. Similarly, the two half-wave reduction potentials of PDI (-0.65 V and -0.90 V vs. Ag/AgCl) were mainly attributed to the formation of its a charge delocalized radical anion [PDI]·- and dianion [PDI]2-, respectively. For TDI with two half-wave reduction potentials at -0.64 V and -1.20 V vs. Ag/AgCl, corresponding to the TDI/[TDI]·- and [TDI]·- /[TDI]2-, which consists well with the two-step reduction reaction of the NDI and PDI. As previous reports [17], the most remarkable feature of their solution-state cyclic voltammograms is possessing the same amount of redox peaks and similar redox peaks shape, indicating to they have similar electrochemical redox states using Ag/AgCl as reference electrode in a dilute solution.

|
Download:
|
| Fig. 1. (a) UV-vis absorptions of NDI, PDI and TDI in DCM at room temperature, and their solution cyclic voltammograms (CVs) recorded (scan rate 50 mV/s) using a glassy carbon electrode (b): all of the experiments were performed at 298 K in argon-purged DCM (1 mmol/L in DCM). | |
{kind=link}
Subsequently, the electrochemical performances of the NDI, PDI and TDI in lithium-ion batteries have been examined initially. As shown in Fig. 2, the CV experiment with the scan rate of 0.1 mV/s was firstly performed to check their electrochemical behaviors. Fig. 2a shows clear reduction potentials the solid-state CV curves of the NDI cathodes in the voltage window of 1.8–3.2 V, exhibiting two pairs of reversible redox peaks centered at 2.31 V and 2.54 V during the first cycle, and this redox process was consistent with the peak corresponding to the reduction of solution-state CV results. Interestingly, solid-state CV curves of the PDI and TDI batteries only reveal (Fig. 2a) one set of peaks, centered at 2.57 and 2.54 V, corresponding to the one two-electron redox steps for the PDI and TDI, respectively. These experimental results were contrary to its reversible electrochemical behavior in solutions, which might be attributed to the unstable species of radical anions with negligible energy barriers in the solid states and thus can be directly transformed into their dianionic species. Encouraged by the interesting CV results obtained for three aromatic diimides batteries, the galvanostatic charge/discharge curves of the NDI, PDI and TDI electrodes at 1.0 C have also been investigated, and their 5th charge/discharge curves were shown in Fig. 2b. The charge/discharge curves of the NDI are composed of two distinctly aclinic voltage plateaus centered at 2.58/2.47 V and 2.35/2.24 V, which indicate two successive electronic redox processes. Notably, unlike the NDI, the PDI showed only one clearly aclinic charge/discharge voltage plateaus centered at 2.61/2.47 V, suggesting that one step reduction of the aromatic diimides carbonyl moiety. During the charge/discharge processes, the TDI displays one-step distorted voltage plateaus mainly centered at 2.80/2.71 V and 2.63/2.46 V. Accordingly, three homologues charge/discharge voltage plateaus are in agreement with the redox peaks in the CV curves, as shown in Fig. 2a, respectively. The dissimilar redox peaks of solid-state CV suggested that three aromatic diimides possess different electronic redox mechanisms, which may affected by the stabilities of their redox intermediates during battery operation. The impact of the differently vertical π-extension on the electronic redox mechanisms of aromatic diimides will be explored further vide infra.

|
Download:
|
| Fig. 2. Cyclic voltammograms of NDI, PDI and TDI batteries in DOL/DME (1:1 in volume) with 1.0 mol/L LiTFSI at room temperature (a), scanned at 0.1 mV/s and a voltage range of 3.2-1.8 V; their galvanostatic charge/discharge curves at a current density of 1.0 C (b); their long-term cycling performance at 1.0 C (c); as well as their cycling performances at current density of 0.2 C, 0.5 C, 1.0 C, 2.0 C and 5.0 C, respectively (d). | |
{kind=link}
The cycling stability of the NDI, PDI and TDI batteries at 1.0 C is shown in Fig. 2c. The TDI can offer specific capacity of 56.7 mAh/g at 1.0 C during the first cycle, which corresponds to 88.2% of its theoretical specific capacity (64.3 mAh/g), and maintains at 51.7 and 41.6 mAh/g after 20 and 100 cycles, respectively. However, the specific capacity of the NDI and PDI batteries delivers 46.6 mAh/g and 44.8 mAh/g at 1.0 C during the first cycle, which corresponds to 50.9% and 59.3% of its theoretical specific capacity (91.5 mAh/g for NDI and 75.5 mAh/g for PDI), respectively. There are obvious capacity losses compared with its theoretical specific capacity, and they only retains a specific capacity of 8.1 mAh/g and 28.6 mAh/g after 100 cycles, respectively. Presumably, this should be attributed to the highly solubility of NDI and PDI than those of TDI in the organic electrolytes, which leads to a drastic loss of the NDI and PDI from the electrodes during charge/discharge process. Furthermore, the rate capability of the NDI, PDI and TDI batteries was measured by cycling successively at different current rates of 0.2 C, 0.5 C, 1.0 C, 2.0 C and 5.0 C, and delivering capacities of 66.6, 19.6, 8.9, 4.6 and 2.2 mAh/g for NDI, and offering capacities of 61.5, 32.1, 28.7, 24.8 and 21.2 mAh/g for PDI, and providing capacities of 61.5, 44.4, 40.2, 37.3 and 34.5 mAh/g for TDI, respectively, as shown in Fig. 2d. Therefore, the TDI battery presented better stable cycling performance and excellent rate capability than those of NDI and PDI batteries.
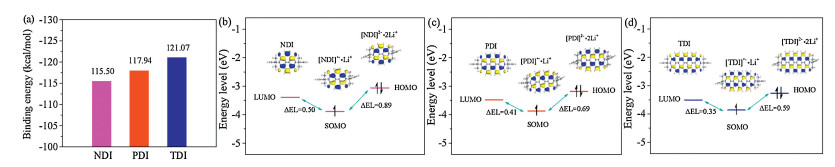
The distinct differences between the charge/discharge voltage plateaus and cycling stabilities of NDI, PDI and TDI, explored experimentally motivated us to get more insights into the effects on the vertical π-extension on electronic redox mechanisms of three aromatic diimides. Their structural optimization, as well as highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energy levels have been performed by the density function theory calculations [18]. The calculations have been implemented without considering the influence of electrolyte solvents, although this can also reproduce the main trends of experimental results. As shown in Fig. 3, a higher LUMO energy level (-5.50 eV) of TDI compared to those of NDI (-6.46 eV) and PDI (-6.01 eV) have been observed, which implies a higher discharge operating voltage. In addition, the energy gaps (Eg) of the HOMO-LUMO for three aromatic diimides have also been obtained, which are 3.07, 2.53 and 1.99 eV for NDI, PDI and TDI, respectively. These values of the HOMO-LUMO energy gaps are consistent with those of the optical energy-band gaps. TDI with a small Eg value favors better electron transfer during electrochemical redox processes, leading to its higher special capacity utilization and rate performance than those of NDI and PDI [19].

|
Download:
|
| Fig. 3. HOMO-LUMO plots of NDI, PDI and TDI and the correlated energy-level diagram, in which the HOMO-LUMO energy gaps are presented. | |
{kind=link}
In order to better understand the energy changes of three aromatic diimides in the different extents of redox properties, we have performed their respective carbonyl reduction states containing one-electron or two-electron reduction per aromatic diimide unit at the B3LYP/6-31 G(d) level. Computational details are described in the Supporting information. We have also calculated their binding energies (BEs), which have been considered to the energies released upon creation of the diimide-Li complexes. As shown in Fig. 4a, the BEs of three aromatic diimides are -115.50, -117.94 and -121.07 kcal/mol for NDI, PDI and TDI, respectively, which indicated that TDI is more favorable binding to lithium atom than those of NDI and PDI [20]. In addition, the stabilities of different redox states of three aromatic diimides have been verified by HOMO or singly occupied molecular orbital (SOMO) energies. As shown in Figs. 4b-d, all the HOMOs/SOMOs of three aromatic diimides in different redox states are well-localized and retained within the structures, which implying that the backbones of three aromatic diimides molecules are able to support one or two negative charges. More importantly, the LUMO–SOMO–HOMO energy levels (ΔEL, 0.50 and 0.83 eV) for NDI with neutral, one or two Li+ electrochemical lithiation fits (Fig. 4b) two-step equivalent well-resolved charge/discharge profile from galvanostatic measurements. More importantly, the theoretical studies speculated that the electrochemical lithiation and delithiation reactions of NDI with large ΔEL differs from those of PDI (Fig. 4c) and TDI (Fig. 4d), reflecting main electrochemical characteristics of the experimental results. Accordingly, the calculated small difference between the first and the second ΔEL is only 0.41 and 0.69 eV for PDI as well as 0.35 and 0.59 eV for TDI, respectively, which answers for the one-step well-resolved charge/discharge profile for PDI but distorted charge/discharge profile for TDI. Also, the small ΔEL of TDI combination with two lithium ions was observed, which also agrees with the high electronical conductivity as well as high operating voltages of TDI in OLIBs [21].

|
Download:
|
| Fig. 4. (a) The calculated Li-binding energies of NDI, PDI and TDI after two-electron reduction complexes; (b) LUMO, SOMO and HOMO plots as well as correlated energy level diagram of NDI and its different redox states; (c) LUMO, SOMO and HOMO plots as well as correlated energy level diagram of PDI and its different redox states; (d) LUMO, SOMO and HOMO plots as well as correlated energy level diagram of TDI and its different redox states. | |
{kind=link}
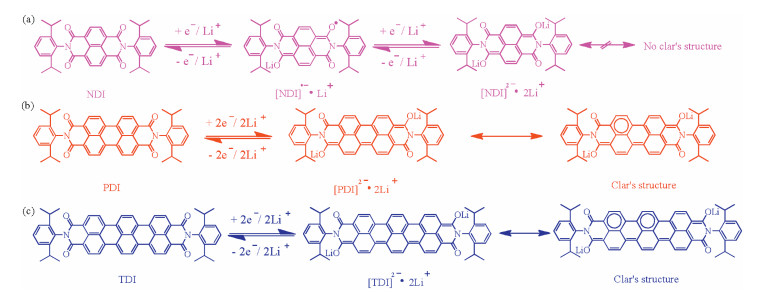
Based on the above investigations, plausible redox mechanisms for three aromatic diimides can be proposed as shown in Scheme 2. For NDI depicted in Scheme 2a, it is supposed to possess a two oneelectron redox mechanism. In first step, Li+ is incorporated into NDI, corresponding to formation of [NDI]·-·Li+, and then another Li+ into the NDI in the second step, corresponding to formation of [NDI]2-·2Li+. However, the one two-electron redox mechanisms are rationalized for PDI and TDI. As depicted in Schemes 2b and c, two Li+ ions are incorporated into PDI and TDI in one step, corresponding to formation of [PDI]2-·2Li+ and [TDI]2-·2Li+, respectively. These redox mechanisms have further been corroborated by their Clar's structures. For [PDI]2-·2Li+, tetracyclic perylene structure possesses one sextet in four rings. However, for hexacyclic terrylene diimide, [TDI]2-·2Li+ has two sextets in six rings.Surprisedly, the dicyclic naphthalene structure of [NDI]2-·2Li+ has no sextet in two rings, which leading to its Clar's structure is not depicted. According to Clar's rule for polycyclic aromatic compounds, the Kekulé resonance structures with maximum benzenoid ring numbers are the most stable [22]. Thus, [TDI]2-·2Li+ with the highest ratio of sextets in rings should be most stable during the electrochemical testing processes.

|
Download:
|
| Scheme 2. Schematic diagram for the proposed reversible redox mechanisms for NDI, PDI and TDI, and depicted their Clar's structure after two-electrode reductions. | |
{kind=link}
In summary, we have demonstrated the impact of on vertical π-extension on the redox mechanisms of aromatic. Different from the laterally π-extended aromatic diimides, the vertical π-extension led to a bathochromic shift of the absorption maxima and different Clar's structures, which is conducive to achieve higher capacity utilizations. Terrylene diimide battery exhibited a high first discharge specific capacity of 56.7 mAh/g at 1.0 C and high initial discharge voltage of 2.70 V, which exceeds those of naphthalene diimide (NDI, 44.6 mAh/g, 2.47 V) and perylene diimide (PDI, 44.8 mAh/g, 2.42 V) batteries. At the same time, TDI battery possesses higher capacity retention than those of NDI and PDI batteries after 100 charge/discharge cycles. More importantly, three vertically π-extended aromatic diimides— NDI, PDI and TDI—exhibited different electronic redox mechanisms with two-step equivalent well-resolved charge/discharge profiles for NDIs, but only one-step well-resolved profile for PDIs, and one-step distorted charge/discharge profile for TDIs when served as cathode materials in lithium-ion batteries. This difference in redox mechanisms has been further rationalized by the chemical stabilities of their reduction states (one-electron as well as two-electron reductions) through density functional theory (DFT) calculations. The HOMO-LUMO energy gap (Eg) and Li-BEs of aromatic diimides confirmed that the redox reaction mechanisms of aromatic diimides are significantly dependent on their vertical π-extension. Accordingly, our research offers an effective strategy for high performance diimide based organic storage energy materials.
AcknowledgmentsThis work is supported by the National Natural Science Foundation of China (No. 21572032), Program for New Century Excellent Talents in Fujian Province University, Natural Science Foundation of Fujian Province (Nos. 2018J01431 and 2018J01690), the Foundation of Science and Technology on Sanming Institute of Fluorochemical Industry (No. FCIT201706GR) as well as Excellent Youth Exchange Program of China Association for Science and Technology in 2017.
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.05.040.
| [1] |
(a) M. Armand, J.M. Tarascon, Nature 451 (2008) 652-657; (b) M. Li, J. Lu, Z. Chen, et al., Adv. Mater. 30 (2018) 1800561; (c) Y. Tang, Y. Zhang, W. Li, et al., Chem. Soc. Rev. 44 (2015) 5926-5940. |
| [2] |
(a) M. Zhu, Y. Huang, Y. Huang, et al., Adv. Funct. Mater. 26 (2016) 4481-4490; (b) K. Sharma, K. Pareek, R. Rohan, et al., Angew. Chem. Int. Ed. 43 (2019) 604-611; (c) W. Li, F. Gao, X. Wang, et al., Angew. Chem. Int. Ed. 55 (2016) 9196-9201; (d) H. Zhang, X. Zhang, Y. Ma, Electrochim. Acta 184 (2015) 347-355. |
| [3] |
(a) Sh. Dai, F. Zhao, Q. Zhang, et al., J. Am. Chem. Soc. 139 (2017) 1336-1343; (b) Z. Zhang, Z. Yang, J. Deng, et al., Small 11 (2015) 675-680; (c) T.B.Song, T.Yokoyama, C.C.Stoumpos, et al., J.Am.Chem.Soc.139 (2017)836-842; (d) Y. Lin, Q. He, F. Zhao, et al., J. Am. Chem. Soc.138 (2016) 2973-2976; (e) H. Imahori, T. Umeyama, S. Ito, Acc. Chem. Res. 42 (2009) 1809-1818. |
| [4] |
(a) Zh. Song, H. Zhou, Energy Environ. Sci. 6 (2013) 2280-2301; (b) M.E. Bhosale, K. Krishnamoorthy, Chem. Mater. 27 (2015) 2121-2126; (c) I.A. Rodríguez-Pérez, Z. Jian, P.K. Waldenmaier, et al., ACS Energy Lett. 1 (2016) 719-723. |
| [5] |
(a) M.Kolek, F.Otteny, P.Schmidt, et al., EnergyEnviron.Sci.10 (2017)2334-2341; (b)J.Winsberg, T.Hagemann, T.Janoschka, et al., Angew.Chem.Int.Ed.55 (2016) 2-28; (c) T. Sun, Z.j. Li, H.g. Wang, et al., Angew. Chem.Int. Ed.55 (2016) 10662-10666. |
| [6] |
(a) L. Chen, S. Liu, L. Zhao, et al., Electrochim. Acta 258 (2017) 677-683; (b) W. Huang, Zh. Zhu, L. Wang, et al., Angew. Chem. Int. Ed. 52 (2013) 9162-9166; (c) Q. Zhao, Zh. Zhu, J. Chen, Adv. Mater. 29 (2017) 1607007; (d) H.g. Wang, Sh. Yuan, Zh. Sia, et al., Energy Environ. Sci. 8 (2015) 3160-3165; (e) H.g. Wang, X.b. Zhang, Chem. -Eur. J. 24 (2018) 18235-18245. |
| [7] |
(a) S.K.M. Nalluri, Zh. Liu, Y. Wu, et al., J. Am. Chem. Soc.138 (2016) 5968-5977; (b) Zh. Luo, L. Liu, Q. Zhao, et al., Angew. Chem. Int. Ed. 56 (2017) 12561-12565. |
| [8] |
(a) D. Chen, A.J. Avestro, Z. Chen, et al., Adv. Mater. 27 (2015) 2907-2912; (b) Ch. Wang, Y. Xu, Y. Fang, et al., J. Am. Chem. Soc. 137 (2015) 3124-3130. |
| [9] |
(a) L. Li, Y.J. Hong, D.Y. Chen, et al., Electrochim. Acta 254 (2017) 255-261; (b) L. Li, Y.J. Hong, D.Y. Chen, et al., Chem. -Eur. J. 23 (2017) 16612-16620; (c) L. Li, H.X. Gong, D.Y. Chen, et al., Chem. -Eur. J. 24 (2018) 13188-13196. |
| [10] |
(a) F. Nolde, J. Qu, Ch. Kohl, et al., Chem. -Eur. J. 11 (2005) 3959-3967; (b) M. Kremer, M. Kersten, Höger S, Org. Chem. Front. 5 (2018) 1825-1829. |
| [11] |
(a) L. Li, Y.J. Hong, Y. Lin, et al., Chem. Commun. 54 (2018) 11941-11944; (b) S. Seifert, K. Shoyama, D. Schmidt, et al., Angew. Chem. Int. Ed. 55 (2016) 6390-6395. |
| [12] |
(a) D. Wu, Z. Xie, Z. Zhou, et al., J. Mater. Chem. A 3 (2015) 19137-19143; (b) J.O. Oña-Ruales, Y. Ruiz-Morales, J. Phys. Chem. A 118 (2014) 12262-12273; (c) A.D. Zdetsis, E.N. Economou, J. Phys. Chem. C 119 (2015) 16991-17003. |
| [13] |
(a) L. Fedélé, F. Sauvage, M. Bécuwe, J. Mater. Chem. A 2 (2014) 18225-18228; (b) H.g. Wang, Sh. Yuan, Zh. Sia, et al., Energy Environ. Sci. 8 (2015) 3160-3165. |
| [14] |
(a) H. Ke, L. Wang, Y. Chen, et al., J. Mol. Catal. A: Chem. 385 (2014) 26-30; (b) M. Marchini, A. Gualandi, L. Mengozzi, et al., Phys. Chem. Chem. Phys. 20 (2018) 8071-8076; (c) F. Nolde, J. Qu, Ch. Kohl, et al., Chem. -Eur. J. 11 (2005) 3959-3967. |
| [15] |
(a) J. Seibt, P. Marquetand, V. Engel, et al., Chem. Phys. 328 (2006) 354-362; (b) M.Chen, Y.J. Bae, C.M. Mauck, et al., J. Am. Chem.Soc.140 (2018) 9184-9192. |
| [16] |
(a) S. Guha, F.S. Goodson, L.J. Corson, et al., J. Am. Chem. Soc. 134 (2012) 13679-13691; (b) F.S. Goodson, D.K. Panda, S. Ray, et al., Org. Biomol. Chem. 11 (2013) 4797-4803; (c) K. Ida, H. Sakai, K. Ohkubo, et al., J. Phys. Chem. C 118 (2014) 7710-7720. |
| [17] |
(a) Y. Wu, M. Frasconi, D.M. Gardner, et al., Angew. Chem. Int. Ed. 53 (2014) 9476-9481; (b) S.T. Schneebeli, M. Frasconi, Zh. Liu, et al., Angew. Chem. Int. Ed. 52 (2013) 13100-13104; (c) Y. Wu, R.M. Young, M. Frasconi, et al., J. Am. Chem. Soc. 137 (2015) 13236-13239. |
| [18] |
(a) Y. Ding, Y. Li, G. Yu, Chemistry 1 (2016) 790-801; (b) Y. Liang, P. Zhang, J. Chen, Chem. Sci. 4 (2013) 1330-1337. |
| [19] |
M. Park, D.S. Shin, J. Ryu, et al., Adv. Mater. 27 (2015) 5141-5146. DOI:10.1002/adma.201502329 |
| [20] |
K.Ch. Kim, T. Liu, S.W. Lee, et al., J. Am. Chem. Soc 138 (2016) 2374-2382. DOI:10.1021/jacs.5b13279 |
| [21] |
(a) D.J. Kim, K.R. Hermann, A. Prokofjevs, et al., J. Am. Chem. Soc. 139 (2017) 6635-6643; (b) T. Ma, Z. Pan, L. Miao, et al., Angew. Chem. Int. Ed. 57 (2018) 3158-3162. |
| [22] |
S. Fujii, T. Enoki, Angew. Chem. Int. Ed. 51 (2012) 7236-7241. DOI:10.1002/anie.201202560 |