 2019, Vol. 30
2019, Vol. 30 


b Cooperative Innovation Center of Lipid Resources and Children's Daily Chemicals, Chongqing University of Education, Chongqing 400067, China;
c School of Chemistry and Chemical Engineering, Chongqing University, Chongqing 401331, China;
d College of Chemistry and Molecular Engineering, Zhengzhou University, Zhengzhou 450006, China
Compounds that incorporate metals have received significant attention in synthetic chemistry, as such compounds can act as catalysts, thereby increasing the reaction rate of a chemical reaction and be recovered unchanged after completion of the reaction [1, 2]. Particularly, the development of metallic-based catalysts has played a major role in the success of the chemical industry. Furthermore, such catalysts play an important role in academia for the synthesis of a series of organic molecules [3-6]. Over the last several decades, late-transition metals have commonly been used as catalysts in organometallic syntheses, which have resulted in the significant advancement of the catalytic activity of coupling reactions and functional group transformations. Numerous studies during the development of organometallic syntheses demonstrated that namely Ru, Rh, Ir, Pd, Pt, Ag, and Au often exhibited unique reactivity that led to numerous metalincorporated compounds being widely applied across the chemical industry and academia [7-16]. Additionally, catalysts containing the aforementioned elements are often relatively expensive, which undoubtedly limits the applications of transition metal catalysis in synthetic chemistry [17-19].
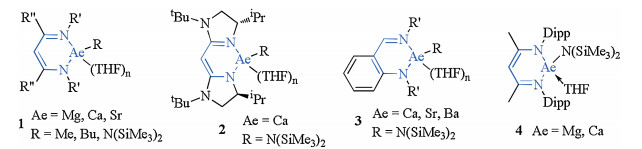
As alternatives to late-transition metal catalysts, alkaline-earth (Ae) metals complexes of Mg, Ca, Sr or Ba have attracted significant attention from synthetic chemists in their pursuit of developing novel synthetic methodologies [20-24]. Ae metals are notable for being inexpensive, as a result of their high natural relative abundance, and having an environmentally benign nature, suggesting that catalytic frameworks, based on these metals, have the potential to be sustainable, economical and green [25-27]. Heteroleptic complexes often need the stabilizing influence of a large multidentate spectator ligand. The ligands for Ae metal precatalysts include: β-diketiminates, borates (bisimino)acenaphthenes, bisimidazolinates, aminotropiminates, triazenides and anilido-imines. A series of Ae metal complexes are summarized in Fig. 1 [28-32].

|
Download:
|
| Fig. 1. Alkaline earth (Ae) metal complexes in organometallic catalysis. | |
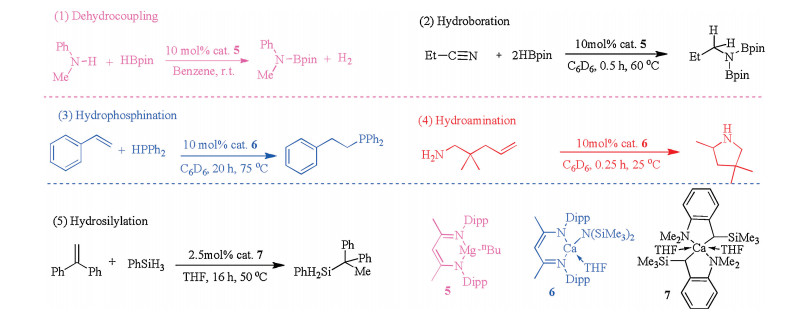
Over the past few decades, an alternative suite of underexploited, and relatively inexpensive, potentially highly sustainable and environmentally benign catalytic compounds, comprising Ae elements in their effectively invariant +2 oxidation state, have been applied to numerous organic synthesis methodologies [20, 33-37]. In particular, the β-diketiminate series of ligands have demonstrated their potential to stabilize low-coordinate and low-valent main-group complexes, especially for Ae metals. Therefore, β-diketiminate-coordinated Ae compounds are the most widely used pre-catalysts. Such catalysts possess strong metal binding abilities and exhibit a broad range of tunable spatial resistance properties. At the outset, β-diketiminate, as a type of stabilizing ligand, is a major component in the catalytic synthesis of polymers, such as the ring-opening polymerization of cyclic esters [38-43]. Recently, Ae metal compounds have been used in various common synthesis methodologies, such as dehydrocoupling, hydroboration, hydrophosphination, hydroamination, and hydrosilylation reactions (Fig. 2) [44-50]. Extensive research on Ae metal compounds has been reported by the Hill group, which concluded that β-diketiminate-coordinated Ae compounds are the effective catalytic component in the above-mentioned reactions [51-56].

|
Download:
|
| Fig. 2. Ae-metal catalyzed organometallic reactions. | |
The valence state of the Ae metals in catalysts is typically 2+. As an example, the catalytic ability of complex 1 (Fig. 1) prepared by Hill et al. was studied in detail. After addition of the pre-catalysts to the reaction, the Ae metals can easily be reduced to Ae metal hydrides. A series of experimental results revealed that the Ae metal hydride complexes generated were the central active species [27, 28, 30, 57, 58]. However, no details are known on the stability and activity of the catalysts. In this work, our group studied the aromaticity of metal heterocycles in β-diketiminate-coordinated Ae compounds to determine if the observed catalytic properties are correlated to aromaticity, followed by discussions on the general properties of the Ae metal hydride compounds.
All calculations were performed using the Gaussian 09 suite of computational programs [59]. Geometry optimizations were performed at the DFT level using the B3-LYP [60-63] hybrid function with a mixed basis set of SDD [64, 65] for Sr and 6–31G(d) [66-71] for other atoms. Harmonic vibrational frequency calculations were performed at the same level to verify the characteristics of all optimized structures, as minimal or transition states, and to derive thermochemical corrections for enthalpy and free energy determinations. In solvent effect of benzene calculations, the M11 [72-74] functional in combination with the 6-311+G(d) basis set (SDD basis set for Sr) was used to calculate the solvation single-point energies to give more accurate energy information. Nucleus-independent chemical shifts (NICS) [75-79] and anisotropy of the induced current density (AICD) [80-82] are used within the computational studies to determine the aromaticity of the β-diketiminate-coordinated Ae compound metal heterocycles. NICS values, for all systems, were obtained by calculating the absolute NMR shielding at the center of the rings and at NICS 1.0 Å above the rings using the GIAO approach at the B3-LYP/6–31G(d) level. NICS values at NICS 1.0 Å above the rings, minimize the paratropic effect and provide a more reliable indication of aromaticity.
We chose the compound 12 as a model to study the stability of the β-diketiminate-coordinated Mg compound, whose ligand was replaced by the hydride. As in the case of aromaticity, magnetic properties can be used as a general descriptor of delocalization. The AICD is a function that is exclusively related to current density paramagnetism, and is determined by the perturbation of the wave function by the magnetic field. Additionally, AICD can be used to measure aromaticity, not only for aromatic systems, but for any conjugation type. The continuous set of gauge transformation (CSGT) method of Bader et al. was applied, which is incorporated within the GAUSSIAN 09 program to calculate current densities.
β-Diketiminate-coordinated Ae compound configurations were optimized at the B3-LYP/6–31G(d) theory level using the Gaussian 09 program, which is shown in Fig. 3 along with the key bond length values. The calculated geometry information, derived from the density functional theory (DFT), is in good agreement with experimental observations by single-crystal analysis. The calculation results predict that each optimized structure has two C—C bonds of the same length. A similar outcome was observed for the C–N and N-Ae bonds in each β-diketiminate-coordinated Ae compound. Furthermore, the length of each C—C or C–N bond is presented as the average of the corresponding single and double bonds, which indicate that each β-diketiminate-coordinated Ae compound can be derived from both the two resonance structures, shown in Fig. 4. Additionally, the length of the N-Ae bond in the ring increases as a function of the metal element content. These observations can be attributed to the fact that the metal atomic radius increases as the period of the metal element increases. Because there are no data relating to N-Ae double bonds, determining if the metal heterocycles, comprising the β-diketiminate-coordinated Ae compounds, are aromatic is difficult. However, the organic region of these compounds is delocalized.

|
Download:
|
| Fig. 3. Optimized structures of β-diketiminate-coordinated Ae compounds. Lengths of key bonds are given in ångstroms. | |

|
Download:
|
| Fig. 4. Two resonance structures for β-diketiminate-coordinated Ae compounds. | |
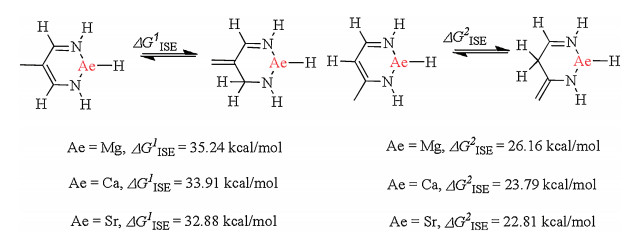
The stability of conjugated π-cyclic systems is indicated by the isomerization energy between the exocyclic isomer and the endocyclic one. Two different isomerization energy calculation types, for the β-diketiminate-coordinated Ae compounds, are shown in Fig. 5. Isomerization results in a non-conjugated π-cyclic system, however, the sum of the π-electrons remains. The calculated results show that the isomerization energy values are >20 kcal/mol. The high isomerization energy values of the β-diketiminate-coordinated Ae compounds are because of the delocalization of the ligand electron. Such high isomerization energies clearly demonstrate that the conjugated π-cyclic system significantly increased the stability of those compounds.

|
Download:
|
| Fig. 5. Isomerization energy calculations for β-diketiminate-coordinated Ae compounds. The relative free energies (ΔG) in benzene are given in kcal/mol. | |
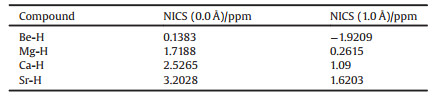
As shown in Fig. 6, in a typical β-diketiminate-coordinated Ae compound, the organic region provides six π electrons. In the presence of an unoccupied Ae p orbital, such compounds form a closed six-orbital-six-electron π system. Therefore, the aromaticity of β-diketiminate-coordinated Ae compounds should be considered to explain the enhanced stability observed. However, bond length equalization should not be the only criterion to determine aromaticity because not all bond-equalized systems are aromatic. A magnetic criterion, namely NICS, was studied to determine the aromaticity of β-diketiminate-coordinated Ae compounds because of its simplicity and efficiency. The NICS values calculated at the center of the rings (NICS (0.0 Å)) and 1.0 Å above the rings (NICS (1.0 Å)), obtained using the gauge invariant atomic orbital (GIAO) procedure at the B3LYP/6–31G(d) level, are given in Table 1. A negative NICS value indicates aromaticity, alternatively a positive value indicates anti-aromaticity. Additionally, a near-zero value indicates non-aromaticity. All absolute NICS values shown in Table 1 are < 4.0 ppm, which clearly demonstrates that all of these compounds exhibit non-aromaticity. Furthermore, the calculated NICS 0.0 Å values are slightly higher than the corresponding NICS 1.0 Å values, which also validates the nonaromaticity character of the β-diketiminate-coordinated Ae compounds.

|
Download:
|
| Fig. 6. Structure of a typical β-diketiminate-coordinated Ae compound. | |
|
|
Table 1 Nucleus-independent chemical shifts (NICS) values for β-diketiminate-coordinated Ae compounds. All NICS values are given in ppm. |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
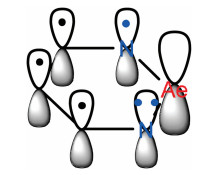
The AICD can be interpreted as a representation of delocalized electrons that provides a simple way for the quantification of conjugative effects and can be used forany type of conjugation (not only aromaticity). Herein, AICD is used to investigate the aromaticity of β-diketiminate-coordinated Ae compounds. 'Left-hand rule' is used to determine current direction induced by a magnetic field in a conductor. If no currents are induced throughout the ring, the system is non-aromatic.
The AICD isosurface of the metallic heterocyclic rings are shown in Fig. 7. The current density vectors plotted on top of the isosurface indicate a weak diamagnetic ring current, which infers a degree of electron delocalization in this area, even though the delocalization is not strong. Additionally, the ring current density of the β-diketiminate-coordinated Ae compound metal heterocycles increases as a function of Ae metal radii. The results suggest that the elemental periodic law influences the induced current density of the β-diketiminate-coordinated Ae compound metal heterocycles. Notably, none of the molecules studied exhibit induced currents that surround the entire metal heterocycle, demonstrating that the metal heterocycles of the four β-diketiminate-coordinated Ae compounds are non-aromatic. We consider that only non-aromaticity complex has free p orbital to interact with amines, because the p orbital of Mg can accept electrons easily. Therefore, we think non-aromaticity can improve the catalytic activities of those compounds.

|
Download:
|
| Fig. 7. Cyclic plane-induced current density map of β-diketiminate-coordinated Ae compound metal heterocycles. | |
{kind=link}
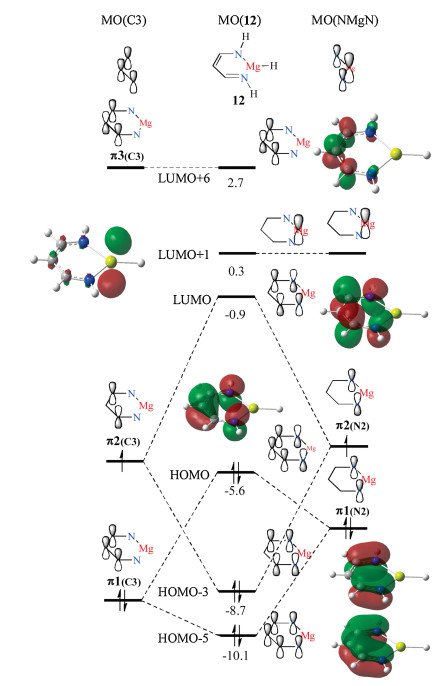
To further elucidate the stability of β-diketiminate-coordinated Ae compounds, a molecular orbital analysis of a model compound 12, is shown in Fig. 8. The molecular orbital analysis for the model β-diketiminate-coordinated Mg hydride is given in the center of Fig. 8, which clearly represents a delocalized character of the π electrons. Both the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) receive contribution from the p orbitals from all carbon and nitrogen atoms. The π electrons of compound 12 are composed of two parts, which involve three carbon moieties (left side) and a N-Mg-N moiety (right side). The combination of the C3 and N-Mg-N fragment orbitals result in the unique molecular orbital of the β-diketiminate-coordinated Mg hydride compound 12. As shown on the left side of Fig. 8, there are three fragment orbitals corresponding to the C3 moiety, which are attributed to π1(C3), π2(C3), and π3(C3), representing bonding, non-bonding, and antibonding orbitals, respectively. In the right side of Fig. 8, the two atomic orbitals, derived from the nitrogen atoms, are referred to as π1(N2) and π2(N2). Furthermore, the p orbital of the Mg atom is isolated and does not interact with other orbitals. The combination of π1(C3) and π1(N2) leads to the formation of the HOMO-5 and HOMO molecular orbitals with energy levels of -10.1 eV and -5.6 eV, respectively. π2(C3) and π2(N2) are of the same symmetry, therefore, their combination can result in the HOMO-3 and LUMO molecular orbitals in compound 12. The energy level of HOMO is 3.1 eV higher than that of HOMO-3, demonstrating that the HOMO and LUMO for compound 12 derive from different symmetry fragments. Additionally, in the molecular orbital analysis, the p orbital of the Mg atom is almost isolated and does not interact with other carbon and nitrogen atoms, therefore, non-aromaticity is observed theoretically.

|
Download:
|
| Fig. 8. Molecular orbitals of β-diketiminate-coordinated Mg compounds. The values under horizontal lines are the orbital levels in eV. | |
{kind=link}
In conclusion, β-Diketiminate-coordinated Ae compounds are a series of catalytically active molecules, which are widely applied in homogeneous organometallic catalysis. Such compounds comprise an Ae metal-incorporated six-membered close-conjugative cycle fitted with six π electrons. Both theoretical and experimental studies were in agreement regarding the nature of the C—C and C–N bonds, in that each of these compounds exhibit a similar equivalency between that of typical single and double bonds. Theoretical NICS and AICD studies demonstrate that the Ae metalincorporated six-membered rings are non-aromatic. Molecular orbital analyses also proved this point. The non-aromaticity of β-diketiminate-coordinated Ae compounds can be attributed to the high-energy p orbital level of the Ae metal, leading to noninteraction with other carbon and nitrogen atoms. Furthermore, DFT calculations revealed relatively high isomerization energies for β-diketiminate-coordinated Ae compounds, which contribute to their stability. The enhanced stability can be explained by p orbital delocalization of the β-diketiminate ligand. Thus, theoretical calculations are demonstrated to be of use to further design stable ligands for Ae-catalyzed organometallic reactions.
AcknowledgmentsThis project was supported by the National Natural Science Foundation of China (Nos. 21822303, 21772020), the Basic and Frontier Research Project of Chongqing Science and Technology Commission (Nos. cstc2018jcyjAX0827), the Project of Science and Technology Collaborative Innovation Platform Construction of Chongqing University of Education (No. 2017XJPT01), the Project of Scientific and Technological Research Program of Chongqing Municipal Education Commission (No. KJQN201801603) and the Cultivation for National Science Foundation of Chongqing University of Education (No.18GZKP01). This project was funded by Children's Research Institute of National Center for Schooling Development Programme and Chongqing University of Education (No. CRIKT201909) and the Fundamental Research Funds for the Central Universities (Chongqing University, No. 2018CDPTCG0001/4).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.06.001.
| [1] |
B.B. Corson, J. Chem. Educ. 24 (1947) 99-103. DOI:10.1021/ed024p99 |
| [2] |
E.C. Ashby, H.Y. Simon, G.B. Robert, J. Am. Chem. Soc. 28 (1970) 435-436. |
| [3] |
H.A.M.L. Stefan, R. Gramage-Doria, B.D. Bruin, et al., Chem. Soc. Rev. 44 (2015) 433-448. DOI:10.1039/C4CS00192C |
| [4] |
Y.Y. Li, Y.J. Wang, L. Zhu, et al., Chin. J. Org. Chem. 39 (2019) 38-46. DOI:10.6023/cjoc201810020 |
| [5] |
Z.H. Shao, H.B. Zhang, Chem. Soc. Rev. 38 (2009) 2745-2755. DOI:10.1039/b901258n |
| [6] |
Y.S. Bai, A. Chotera, O. Taran, et al., Chem. Soc. Rev. 47 (2018) 5444-5456. DOI:10.1039/C8CS00174J |
| [7] |
C.H. Shan, L. Zhu, L.B. Qu, et al., Chem. Soc. Rev. 47 (2018) 7552-7576. DOI:10.1039/C8CS00036K |
| [8] |
Z.J. Kuang, H.H. Chen, J.X. Yan, et al., Org. Lett. 20 (2018) 5153-5157. DOI:10.1021/acs.orglett.8b02077 |
| [9] |
C.H. Shan, K.B. Zhong, X.T. Qi, et al., Org. Chem. Front. 5 (2018) 3178-3185. DOI:10.1039/C8QO00699G |
| [10] |
S.S. Liu, K. Kang, S.H. Liu, et al., Organometallics 37 (2018) 3901-3908. DOI:10.1021/acs.organomet.8b00579 |
| [11] |
X.T. Qi, X.F. Liu, L.B. Qu, et al., J. Catal. 362 (2018) 25-34. DOI:10.1016/j.jcat.2018.03.016 |
| [12] |
Y.X. Luo, S. Liu, D.D. Xu, et al., J. Organomet. Chem. 864 (2018) 148-153. DOI:10.1016/j.jorganchem.2018.03.016 |
| [13] |
Y.Z. Li, L.F. Zou, R.P. Bai, et al., Org. Chem. Front. 5 (2018) 615-622. DOI:10.1039/C7QO00850C |
| [14] |
L. Zhu, J.H. Ye, M. Duan, et al., Org. Chem. Front. 5 (2018) 633-639. DOI:10.1039/C7QO00838D |
| [15] |
S. Liu, X.T. Qi, L.B. Qu, et al., Catal. Sci. Technol. 8 (2018) 1645-1651. DOI:10.1039/C7CY02367G |
| [16] |
S. Liu, T. Zhang, L. Zhu, et al., Chem. Commun. 54 (2018) 13551-13554. DOI:10.1039/C8CC08335E |
| [17] |
G.E. Dobereiner, R.H. Crabtree, Chem. Rev. 110 (2010) 681-703. DOI:10.1021/cr900202j |
| [18] |
R. Rochat, M.J. Lopez, H. Tsurugi, et al., Chem. Cat. Chem. 8 (2016) 10-20. |
| [19] |
R.J. Schwamm, B.M. Day, N.E. Mansfield, et al., Dalton Trans. 43 (2014) 14302-14314. DOI:10.1039/C4DT01097C |
| [20] |
P.J. Bailey, C.M.E. Dick, S. Fabre, et al., J. Chem. Soc., Dalton Trans. 10 (2000) 1655-1661. |
| [21] |
S.J. Bonyhady, S.P. Green, C. Jones, et al., Angew. Chem. Int. Ed. 48 (2009) 2973-2977. DOI:10.1002/anie.200900331 |
| [22] |
S.P. Green, C. Jones, A. Stasch, Angew. Chem. Int. Ed. 47 (2008) 9079-9083. DOI:10.1002/anie.200803960 |
| [23] |
M. Arrowsmith, W.M.S. Shepherd, S.M. Hill, et al., Chem. Commun. 50 (2014) 12676-12679. DOI:10.1039/C4CC05223D |
| [24] |
(a) S. Harder, Chem. Rev. 110 (2010) 3852-3876; (b) M.S. Hill, D.J. Liptrot, C. Weetman, Chem. Soc. Rev. 45 (2016) 972-988; (c) M. Arrowsmith, M.S. Hill, G. Kociok-Köhn, Chem. -Eur. J. 21 (2015) 10548-10557. |
| [25] |
A. Yaroshevsky, Geochem. Int. 44 (2006) 48-55. DOI:10.1134/S001670290601006X |
| [26] |
D.D. Xu, C.H. Shan, R.P. Bai, et al., J. Org. Chem. 37 (2017) 1231-1236. |
| [27] |
Y.Y. Li, Y.H. Cheng, C.H. Shan, et al., Chin. J. Org. Chem. 38 (2018) 1885-1896. DOI:10.6023/cjoc201804031 |
| [28] |
S.P. Green, C. Jones, Science 318 (2007) 1754-1757. DOI:10.1126/science.1150856 |
| [29] |
A. Stasch, C. JonesStable, Dalton Trans. 40 (2011) 5659-5672. DOI:10.1039/c0dt01831g |
| [30] |
S. Harder, Alkaline-Earth Metal Compounds: Oddities and Applications, Springer, Berlin Heidelberg, 2013.
|
| [31] |
M.S. Hill, D.J. Liptrot, C. Weetman, et al., Chem. Soc. Rev. 45 (2016) 972-988. DOI:10.1039/C5CS00880H |
| [32] |
M.T. Ma, W.F. Wang, W.W. Yao, et al., Chin. J. Org. Chem. 36 (2016) 72-82. DOI:10.6023/cjoc201507015 |
| [33] |
M. Arrowsmith, M.R. Crimmin, M.S. Hill, et al., Dalton Trans. 43 (2014) 14249-14256. DOI:10.1039/C3DT53542H |
| [34] |
C.X. Cui, Y. Tian, Y.P. Zhang, et al., Carbon 143 (2019) 3859390. |
| [35] |
B. Liu, T. Roisnel, J.F. Carpentier, et al., Angew. Chem. Int. Ed. 51 (2012) 4943-4946. DOI:10.1002/anie.201200364 |
| [36] |
C.X. Cui, C.H. Shan, Y.P. Zhang, et al., Chem. -Asian J. 13 (2018) 1076-1088. DOI:10.1002/asia.201800146 |
| [37] |
H.H. Chen, L. Zhu, K.B. Zhong, et al., Chin. Chem. Lett. 29 (2018) 1237-1241. DOI:10.1016/j.cclet.2018.03.018 |
| [38] |
Z.Y. Yu, Z.C. Jin, M. Duan, et al., J. Org. Chem. 83 (2018) 9729-9740. DOI:10.1021/acs.joc.8b01259 |
| [39] |
A. Rossin, M. Peruzzini, et al., Chem. Rev. 116 (2016) 8848-8872. DOI:10.1021/acs.chemrev.6b00043 |
| [40] |
M.H. Chisholm, N.W. Eilerts, et al., Chem. Commun. 7 (1996) 853-854. |
| [41] |
M.H. Chisholm, N.W. Eilerts, J.C. Huffman, et al., J. Am. Chem. Soc. 122 (2000) 11845-11854. DOI:10.1021/ja002160g |
| [42] |
M.H. Chisholm, J.C. Huffman, K. Phomphrai, et al., J. Chem. Soc. Dalton Trans. 3 (2001) 222-224. |
| [43] |
B.J. O'Keefe, M.A. Hillmyer, W.B. Tolman, et al., J. Chem. Soc. Dalton Trans. 15 (2001) 2215-2224. |
| [44] |
D.J. Liptrot, M.S. Hill, M.F. Mahon, et al., Angew. Chem. Int. Ed. 54 (2015) 13362-13365. DOI:10.1002/anie.201505949 |
| [45] |
C. Weetman, M.D. Anker, M. Arrowsmith, et al., Chem. Sci. 7 (2016) 628-641. DOI:10.1039/C5SC03114A |
| [46] |
M.R. Crimmin, A.G.M. Barrett, M.S. Hill, et al., Organometallics 26 (2007) 2953-2956. DOI:10.1021/om070200k |
| [47] |
M.R. Crimmin, I.J. Casely, M.S. Hill, J. Am. Chem. Soc. 127 (2005) 2042-2043. DOI:10.1021/ja043576n |
| [48] |
F. Buch, J. Brettar, S. Harder, Angew. Chem. Int. Ed. 45 (2006) 2741-2745. DOI:10.1002/anie.200504164 |
| [49] |
J. Wang, D.M. Cui, Chin. J. Org. Chem. 36 (2016) 1163-1183. DOI:10.6023/cjoc201512010 |
| [50] |
D.D. Xu, C.H. Shan, Y.Z. Li, et al., Inorg. Chem. Front. 4 (2017) 1813-1820. DOI:10.1039/C7QI00459A |
| [51] |
M.H. Chisholm, C. Gallucci, K. Phomphrai, Inorg. Chem. 43 (2004) 6717-6725. DOI:10.1021/ic0490730 |
| [52] |
M. Arrowsmith, M.S. Hill, T. Hadlington, et al., Organometallics 30 (2011) 5556-5559. DOI:10.1021/om2008138 |
| [53] |
M. Arrowsmith, T.J. Hadlington, M.S. Hill, et al., Chem. Commun. 48 (2012) 4567-4569. DOI:10.1039/c2cc30565h |
| [54] |
D. Mukherjee, A. Ellern, A.D. Sadow, Chem. Sci. 5 (2014) 959-964. DOI:10.1039/C3SC52793J |
| [55] |
N.L. Lampland, M. Hovey, D. Mukherjee, et al., ACS Catal. 5 (2015) 4219-4226. DOI:10.1021/acscatal.5b01038 |
| [56] |
M. Arrowsmith, M.S. Hill, G. Kociok-Köhn, Chem. -Eur. J. 19 (2013) 2776-2783. DOI:10.1002/chem.201203190 |
| [57] |
C. Weetman, M.S. Hill, M.F. Mahon, Chem. -Eur. J. 22 (2016) 7158-7162. DOI:10.1002/chem.201600681 |
| [58] |
J. Intemann, M. Lutz, S. Harder, Organometallics 33 (2014) 5722-5729. DOI:10.1021/om500469h |
| [59] |
M.J. Frisch, G.W. Trucks, H.B. Schlegel, et al., Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, 2013.
|
| [60] |
C.T. Lee, W.T. Yang, G.P. Robert, Phys. Rev. B 37 (1988) 785-789. DOI:10.1103/PhysRevB.37.785 |
| [61] |
A.D. Becke, J. Chem. Phys. 98 (1993) 5648-5652. DOI:10.1063/1.464913 |
| [62] |
P.J. Stephens, F.J. Devlin, C.F. Chabalowski, et al., J. Phys. Chem. 98 (1994) 11623-11627. DOI:10.1021/j100096a001 |
| [63] |
C. Adamo, J. Chem. Phys. 110 (1999) 6158-6170. DOI:10.1063/1.478522 |
| [64] |
M. Dolg, U. Wedig, H. Stoll, et al., J. Chem. Phys. 86 (1987) 866-872. DOI:10.1063/1.452288 |
| [65] |
P. Schwerdtfeger, M. Dolg, W.H.E. Schwarz, et al., J. Chem. Phys. 91 (1989) 1762-1774. DOI:10.1063/1.457082 |
| [66] |
T.A. Keith, R.F.W. Bader, J. Chem. Phys. 99 (1993) 3669-3682. DOI:10.1063/1.466165 |
| [67] |
T.A. Keith, R.F.W. Bader, Chem. Phys. Lett. 194 (1992) 1-8. DOI:10.1016/0009-2614(92)85733-Q |
| [68] |
C.X. Cui, D.D. Xu, B.W. Ding, et al., J. Comput. Chem. 40 (2019) 657-670. DOI:10.1002/jcc.25752 |
| [69] |
N.H. Martin, D.M. Loveless, K.L. Main, et al., J. Mol. Graph. Model. 25 (2006) 389-395. DOI:10.1016/j.jmgm.2006.02.006 |
| [70] |
W.J. Hehre, R. Ditchfield, J.A. Pople, J. Chem. Phys. 56 (1972) 2257-2261. DOI:10.1063/1.1677527 |
| [71] |
M.M. Francl, W.J. Pietro, W.J. Hehre, et al., J. Chem. Phys. 77 (1982) 3654-3665. DOI:10.1063/1.444267 |
| [72] |
R. Peverati, D.G. Truhlar, J. Phys. Chem. Lett. 2 (2011) 2810-2817. DOI:10.1021/jz201170d |
| [73] |
Y. Xiong, Z.Z. Du, H.H. Chen, et al., J. Am. Chem. Soc. 141 (2019) 961-971. DOI:10.1021/jacs.8b10939 |
| [74] |
Y.Y. Li, M.J. Wu, H.H. Chen, et al., Front. Chem. 7 (2019) 149. DOI:10.3389/fchem.2019.00149 |
| [75] |
J.R. Cheeseman, G.W. Trucks, T.A. Keith, et al., J. Chem. Phys. 104 (1996) 5497-5509. DOI:10.1063/1.471789 |
| [76] |
P.V.R. Schleyer, C. Maerker, A. Dransfeld, et al., J. Am. Chem. Soc. 118 (1996) 6317-6318. DOI:10.1021/ja960582d |
| [77] |
A. Saieswari, U.D. Priyakumar, G.N. Sastry, J. Mol. Struc. (Theochem.) 663 (2003) 145-148. DOI:10.1016/j.theochem.2003.08.130 |
| [78] |
S. Patchkovskii, W. Thiel, J. Mol. Model. 6 (2000) 67-75. DOI:10.1007/PL00010736 |
| [79] |
Z.F. Chen, C.S. Wannere, Chem. Rev. 105 (2005) 3842-3888. DOI:10.1021/cr030088+ |
| [80] |
D. Geuenich, K. Hess, F. Kohle, et al., Chem. Rev. 105 (2005) 3758-3772. DOI:10.1021/cr0300901 |
| [81] |
R. Herges, D. Geuenich, J. Phys. Chem. A 105 (2001) 3214-3220. DOI:10.1021/jp0034426 |
| [82] |
D. Quinonero, C. Garau, A. Frontera, et al., Chem. -Eur. J. 8 (2002) 433-438. DOI:10.1002/1521-3765(20020118)8:2<433::AID-CHEM433>3.0.CO;2-T |