 2019, Vol. 30
2019, Vol. 30 


b State Key Laboratory of Environmental Chemistry and Ecotoxicology, Research Center for Eco-Environmental Sciences, Chinese Academy of Sciences, Beijing 100085, China;
c Laboratory of Environmental Nanotechnology and Health Effect, Research Center for Eco-Environmental Sciences, Chinese Academy of Sciences, Beijing 100085, China;
d Department of Chemistry and Biochemistry, Florida International University, Miami, FL 33199, United States
The wide occurrence of legacy and emerging contaminants in various environmental compartments has draw great concern in recent decades. Fortunately, naturally biological and chemical degradation contributes for the attenuation of these contaminants in the environment. The highly active hydroxyl radical (·OH) with a standard potential 2.8 V plays a critical role in the chemical degradation of various hazardous compounds due to its strong oxidizing power [1, 2]. It is generally believed that in aquatic environments hydroxyl radical is photochemically produced [3] from dissolved organic matter [4, 5], nitrate/nitrite [6-8], or iron and its complexes [9, 10].
However, in recent years, it was discovered that hydroxyl radical can also form at anoxic-oxic interfaces even in the absence of sunlight [10-12]. The dark formation of hydroxyl radical has been widely observed in soil [13], sediment [11, 12], and surface waters [13]. During oxygenation, reduced substances in anoxic environments could transfer electrons to oxygen to form hydrogen peroxide (H2O2), and then generate hydroxyl radical [13]. In natural environments, the hydroxyl radical formation rates increase with the concentrations of dissolved organic matter (DOM) and reduced iron [13], suggesting the critical role of reduced organic matter and iron as electron donors. Simulation experiments further confirmed the important roles of reduced organic matter [14, 15] and iron minerals [16, 17] in hydroxyl radical formation. Reduced iron minerals, referring to ferrous (Fe2+)-rich minerals, widely present in soil, sediment, and suspended particles in the water column [18-20]. It was discovered that hydroxyl radical could be formed upon dark oxygenation of various reduced iron minerals, including pyrite [16, 21, 22], reduced nontronite [17, 23], and mackinawite [24]. The resulting hydroxyl radical could induce the oxidative degradation and detoxification of trichloroethylene [23], tetracycline [11], and arsenite [24]. It should also be noted that due to the heterogeneous interface generation of hydroxyl radical from the oxygenation of reduced iron minerals, the concentration of hydroxyl radical close to the reduced iron minerals should be much higher than that of the bulk solution, similar to the microheterogeneous radical distributions in irradiated humic acid solutions [25]. Thus, the adsorption of contaminants onto these reduced iron minerals [26, 27] could further increase the reaction probability between them and hydroxyl radical.
In this work, the hydroxyl radical formation upon dark oxidation of various reduced iron minerals, including siderite (FeCO3), magnetite (Fe3O4), pyrite (FeS2), reduced nontronite (ironbearing smectite clay), and mackinawite (FeS), was compared at different oxygen conditions. The interactive effects of mackinawite with environmental factors (pH, Cl- and DOM) were investigated on the formation of hydroxyl radical.
Firstly, the commercial and synthesized iron minerals were characterized by using X-ray diffraction (XRD) (Fig. S1 in Supporting information) and transmission electron microscopy (TEM) (Fig. S2 in Supporting information). The XRD pattern in Figs. S1a-c confirmed the occurrence of crystalline-phased siderite, pyrite, and magnetite, respectively. The XRD pattern of nontronite (Nau-2) (Fig. S1d) was similar to that of smectite, confirming that nontronite is an iron-bearing smectite [28]. Fig. S1e clearly showed that the XRD pattern of synthetic FeS had a characteristic of mackinawite. The TEM of the synthetic mackinawite (Figs. S2a and b) showed that these particles were near-spherical with an average diameter of 5.8 ± 1.7 nm. The high resolution TEM image (Fig. S2c) showed that the distance between two adjacent planes in nanoparticle was 5.09 Å, corresponding to the mackinawite (001) lattice planes [29]. The energy dispersive spectrometry (EDS) shown in Fig. S2d further confirmed that these particles in TEM contained Fe and S.
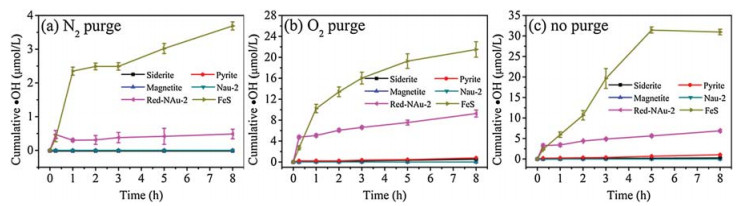
By using the hydroxylation of benzoic acid (BA) to p-hydroxybenzoic acid (p-HBA) as a probe reaction, the cumulative concentrations of ·OH produced from the dark oxidation of various iron minerals (0.483 g/L or 8.65 mmol/L total Fe concentration) were compared quantitatively (Fig. 1). Generally, the cumulative ·OH under anoxic environment (N2 purging) was negligible or at very low concentrations. However, under oxic (O2 purging or no purging) environments, the cumulative ·OH concentrations increased quickly within the initial 1 h, and slowed afterwards for siderite, pyrite, reduced nontronite, and mackinawite. Differently for magnetite and nontronite, regardless of anoxic or oxic conditions, no formation of ·OH was observed. The significant increase of ·OH formation from reduced nontronite than that from nontronite suggests that structural Fe2+ plays a critical role in ·OH formation. The increase of cumulative ·OH under oxic environments also demonstrated the important role of O2 in this process. This is reasonable that structural Fe2+ and O2 act as electron donor and acceptor respectively to produce H2O2 and then to ·OH [23, 24]. The ·OH formation rates were different for the four minerals, decreasing in the following order: mackinawite (30.96 μmol/L) > reduced nontronite (6.87 μmol/L) > pyrite (1.04 μmol/L) > siderite (0.28 μmol/L). It should be noted that the presence of O2 in the system (O2 purging or no purging) has different effects on ·OH formation for different iron minerals. For siderite and reduced Nau-2, purging with O2 increased ·OH formation than without purging. However, for pyrite, O2 purging deceased the ·OH formation than without purging. Interestingly, for mackinawite, O2 purging increased the cumulative ·OH within the first 2 h but decreased afterwards, in comparison to no purging. The differential ·OH formation under various O2 concentrations may be ascribed to the different electron transfer from inter to surface of the iron minerals [17]. We also observed that different iron minerals were produced from mackinawite under varying O2 concentrations. The XRD patterns (Fig. S3 in Supporting information) showed that lepidocrocite (γ-FeO(OH)) and greigite (Fe3S4) were the oxidation products under O2 purging and no purging, respectively. The different oxidation products indicated different transformation pathways, which could also influence the ·OH formation. The oxidation of mackinawite into greigite often involves a solid state transformation process [30]. However, the oxidation of mackinawite into lepidocrocite generally follows a process including dissolution and precipitation of Fe3+ [30]. The formation of ·OH from mackinawite was comparable with that of dissolved Fe2+ (Fig. S4 in Supporting information), suggesting the importance of structural Fe2+ in this process, although the contribution of dissolved Fe2+ is also possible [16]. Considering that reduced iron minerals are abundant and widespread in soils, overlying water, and sediments, this finding is important for understanding the dark formation of ·OH in these environments.

|
Download:
|
| Fig. 1. Cumulative ·OH production upon dark oxidation of various iron minerals (0.483 g/L Fe, pH 6.5) under (a) N2 purge, (b) O2 purge and (c) no purge. | |
{kind=link}
The effect of pH on ·OH production was further investigated by using mackinawite as an iron mineral model (Fig. 2). Cumulative concentrations of ·OH decreased sharply from 62.95 μmol/L to 9.44 μmol/L as the initial pH increased from 3.6 to 8.0. Similarly, it declined from 129.01 μmol/L to 3.87 μmol/L with pH changing from 3.6 to 8.0 under O2 purging. The higher ·OH generation at lower pH is attributed to (1) partial dissolution of FeS particles and thus enhanced release of Fe2+ (Fig. S5 in Supporting information), and (2) decreased ·OH generation efficiency in homo- and heterogeneous Fenton reactions at higher pH [31]. The absence of H+ should inhibit the decomposition of H2O2 and the subsequent production of ·OH [32]. Moreover, the ferrous minerals are deactivated with the formation of ferric oxyhydroxide at higher pH [33].

|
Download:
|
| Fig. 2. The effect of pH on ·OH production by mackinawite under (a) N2 purge, (b) O2 purge and (c) no purge. | |
{kind=link}
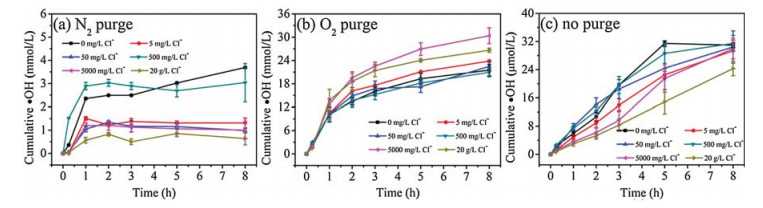
Fig. 3 shows the ·OH production by mackinawite in the presence of varying Cl- concentrations. Generally, in the concentration range of 0–20 g/L, Cl- had a limited effect on ·OH production under O2 purging. However, without purging, the ·OH production kinetics changed with the increasing Cl- concentration. At lower Cl- concentrations, a biphasic ·OH production was observed, generally being quick within the initial 1 h and slowing down afterwards. However, with further increase of Cl- concentration, a pseudo-linear formation kinetics of ·OH was observed, especially at 20 g/L of Cl- concentration. In addition, the increasing of Cl- concentration to 20 g/L also lowered the ·OH production by 27% (from 30 μmol/L to 22 μmol/L). The effect of Cl- can be attributed to (1) the complexation of Cl- and Fe2+/3+ decreases the formation of ·OH [34], and (2) Cl-, as a radical scavenger, decreases the steady state concentration of ·OH [35].

|
Download:
|
| Fig. 3. Effect of Cl- concentration on ·OH production by mackinawite at pH 6.5 under (a) N2 purge, (b) O2 purge and (c) no purge. | |
{kind=link}
The effect of ubiquitous DOM on ·OH formation from mackinawite was probed by using Suwannee River humic acid (SRHA) asa model(Fig. 4). Although DOM could produce ·OH under sunlight irradiation [2], it can also act as a scavenger of this highly reactive radical [36, 37]. Surprisingly, increasing DOM concentration (from 0 to 80 mg/L as total organic carbon) slightly enhanced ·OH formation from 23.92 μmol/L to 31.70 μmol/L without purging, but this enhancement was significant under O2 purging as ·OH formation increased from 14.70 μmol/L to 50.24 μmol/L. Despite that the ·OH formation was lower with O2 purging than no purging in the absence of DOM, O2 purging greatly increased the ·OH formation in the presence of 80 mg/L DOM. This finding is significant considering the ubiquitous co-occurrence of DOM with iron minerals in soil, overlying water, and sediment. Similarly, lowmolecular-weight organic acids (LMWOAs) (e.g., citrate and oxalate) also enhanced the ·OH production from pyrite oxidation [21]. It was proposed that LMWOAs could increase the dissolution of pyrite and the formation and oxidation of Fe2+-LMWOA complexes by O2, which in turn mainly contributed to ·OH production [21]. In addition, the acceleration of pyrite oxidation by Fe3+-LMWOAs could also increase ·OH production [21]. We observed that DOM significantly increased the dissolved Fe2+ and Fe3+ under oxic condition, but not at anoxic condition (Fig. S6 in Supporting information). This result supports that the complexation of Fe2+ or Fe3+ with DOM [38, 39] increased the electron transfer from mackinawite to O2 and then enhanced the ·OH production.

|
Download:
|
| Fig. 4. Effect of humic acid on ·OH production by mackinawite at pH 6.5 under (a) N2 purge, (b) O2 purge and (c) no purge. | |
{kind=link}
In summary, we evaluated and compared the differential ability of various reduced iron minerals (siderite, pyrite, magnetite, reduced nontronite, mackinawite) to produce ·OH and investigated the effect of typical environmental factors on ·OH production from mackinawite oxidation. ·OH production was generally observed from the oxidation of reduced iron minerals, following the order: mackinawite > reduced nontronite > pyrite > siderite. ·OH production increased with decreasing pH, highlighting the importance of this process in acidic environments, e.g., acid mine drainage. Cl- has little effect on ·OH production, indicating this process is still validated in saline environments. More importantly, DOM-enhanced ·OH production highlights the importance of this process in natural environments where DOM is ubiquitous. This sunlight-independent pathway in which ·OH forms during oxidation of reduced iron minerals is important for a complementary understanding of the degradation and transformation of various inorganic and organic pollutants in the redox-fluctuation environments.
AcknowledgmentsThis work is financially supported by the National Natural Science Foundation of China (No. 21777178), Key Projects for Frontier Sciences of the Chinese Academy of Sciences (No. QYZDBSSWDQC018), and the CAS Interdisciplinary Innovation Team (No. JCTD-2018-04). Y. Yin acknowledges supports from the National Young Top-Notch Talents (No. W03070030) and Youth Innovation Promotion Association of the Chinese Academy of Sciences (No. 2016037).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.09.003.
| [1] |
C. Buck, N. Skillen, J. Robertson, P. Robertson, Chin. Chem. Lett. 29 (2018) 773-777. DOI:10.1016/j.cclet.2018.04.022 |
| [2] |
Q. Chen, L. Chen, J. Qi, et al., Chin. Chem. Lett. 30 (2019) 1214-1218. DOI:10.1016/j.cclet.2019.03.002 |
| [3] |
K. Mopper, X.L. Zhou, Science 250 (1990) 661-664. DOI:10.1126/science.250.4981.661 |
| [4] |
M.M. Dong, F.L. Rosario-Ortiz, Environ. Sci. Technol. 46 (2012) 3788-3794. DOI:10.1021/es2043454 |
| [5] |
D. Zhang, S. Yan, W. Song, Environ. Sci. Technol. 48 (2014) 12645-12653. DOI:10.1021/es5028663 |
| [6] |
R.G. Zepp, J. Hoigne, H. Bader, Environ. Sci. Technol. 21 (1987) 443-450. DOI:10.1021/es00159a004 |
| [7] |
O.C. Zafiriou, M.B. True, Mar. Chem. 8 (1979) 33-42. DOI:10.1016/0304-4203(79)90030-6 |
| [8] |
O.C. Zafiriou, M.B. True, Mar. Chem. 8 (1979) 9-32. DOI:10.1016/0304-4203(79)90029-X |
| [9] |
E.M. White, P.P. Vaughan, R.G. Zepp, Aquat. Sci. 65 (2003) 402-414. DOI:10.1007/s00027-003-0675-4 |
| [10] |
D.H. Gonzalez, C.K. Cala, Q. Peng, S.E. Paulson, Environ. Sci. Technol. 51 (2017) 7676-7685. DOI:10.1021/acs.est.7b01299 |
| [11] |
M. Tong, S. Yuan, S. Ma, et al., Environ. Sci. Technol. 50 (2016) 214-221. DOI:10.1021/acs.est.5b04323 |
| [12] |
P. Liao, K. Yu, Y. Lu, et al., Chem. Eng. J. 368 (2019) 700-709. DOI:10.1016/j.cej.2019.03.018 |
| [13] |
S.E. Page, G.W. Kling, M. Sander, et al., Environ. Sci. Technol. 47 (2013) 12860-12867. DOI:10.1021/es4033265 |
| [14] |
S.E. Page, M. Sander, W.A. Arnold, K. McNeill, Environ. Sci. Technol. 46 (2012) 1590-1597. DOI:10.1021/es203836f |
| [15] |
P. Liao, Y. Liang, Z. Shi, ACS Earth Space Chem. 3 (2019) 484-494. DOI:10.1021/acsearthspacechem.8b00181 |
| [16] |
P. Zhang, W. Huang, Z. Ji, C. Zhou, S. Yuan, Geochim. Cosmochim. Acta 238 (2018) 394-410. DOI:10.1016/j.gca.2018.07.018 |
| [17] |
S. Yuan, X. Liu, W. Liao, et al., Geochim. Cosmochim. Acta 223 (2018) 422-436. DOI:10.1016/j.gca.2017.12.025 |
| [18] |
D.R. Lovley, Microbiol. Rev. 55 (1991) 259-287. |
| [19] |
K.A. Weber, L.A. Achenbach, J.D. Coates, Nat. Rev. Microbiol. 4 (2006) 752-764. DOI:10.1038/nrmicro1490 |
| [20] |
D.E. Canfield, Geochim. Cosmochim. Acta 53 (1989) 619-632. DOI:10.1016/0016-7037(89)90005-7 |
| [21] |
P. Zhang, S. Yuan, Geochim. Cosmochim. Acta 218 (2017) 153-166. DOI:10.1016/j.gca.2017.08.032 |
| [22] |
P. Zhang, S. Yuan, P. Liao, Geochim. Cosmochim. Acta 172 (2016) 444-457. DOI:10.1016/j.gca.2015.10.015 |
| [23] |
X. Liu, S. Yuan, M. Tong, D. Liu, Water Res. 113 (2017) 72-79. DOI:10.1016/j.watres.2017.02.012 |
| [24] |
D. Cheng, S. Yuan, P. Liao, P. Zhang, Environ. Sci. Technol. 50 (2016) 11646-11653. DOI:10.1021/acs.est.6b02833 |
| [25] |
D.E. Latch, K. McNeill, Science 311 (2006) 1743-1747. DOI:10.1126/science.1121636 |
| [26] |
M.Y. Ahn, T.R. Filley, C.T. Jafvert, et al., Environ. Sci. Technol. 40 (2006) 215-220. DOI:10.1021/es051415t |
| [27] |
L. Clausen, I. Fabricius, J. Environ. Qual. 30 (2001) 858-869. DOI:10.2134/jeq2001.303858x |
| [28] |
J.R. Cui, Z.P. Zhang, H.W. Yam, China Powder Sci. Technol. 25 (2019) 55-64. |
| [29] |
D. Csakberenyi-Malasics, J.D. Rodriguez-Blanco, V.K. Kis, et al., Chem. Geol. 294 (2012) 249-258. |
| [30] |
Y. Lan, A.S.E. Madden, E.C. Butler, Environ. Sci. Proc. Impacts 18 (2016) 1266-1273. DOI:10.1039/C6EM00461J |
| [31] |
Y. Deng, J.D. Englehardt, Water Res. 40 (2006) 3683-3694. DOI:10.1016/j.watres.2006.08.009 |
| [32] |
C. Walling, Acc. Chem. Res. 8 (1975) 125-131. DOI:10.1021/ar50088a003 |
| [33] |
R.J. Bigda, Chem. Eng. Prog. 91 (1995) 62-66. |
| [34] |
J. de Laat, G.T. Le, B. Legube, Chemosphere 55 (2004) 715-723. DOI:10.1016/j.chemosphere.2003.11.021 |
| [35] |
J. Kiwi, A. Lopez, V. Nadtochenko, Environ. Sci. Technol. 34 (2000) 2162-2168. DOI:10.1021/es991406i |
| [36] |
P. Westerhoff, G. Aiken, G. Amy, J. Debroux, Water Res. 33 (1999) 2265-2276. DOI:10.1016/S0043-1354(98)00447-3 |
| [37] |
D. Zhang, Y. Yin, Y. Li, Y. Cai, J. Liu, Sci. Total Environ. 578 (2017) 535-541. DOI:10.1016/j.scitotenv.2016.10.222 |
| [38] |
C. Catrouillet, M. Davranche, A. Dia, et al., Chem. Geol. 372 (2014) 109-118. DOI:10.1016/j.chemgeo.2014.02.019 |
| [39] |
X. Ou, S. Chen, X. Quan, H. Zhao, J. Geochem. Explor. 102 (2009) 49-55. DOI:10.1016/j.gexplo.2009.02.003 |