 2019, Vol. 30
2019, Vol. 30 


b Key Laboratory of Water and Wastewater Treatment(HUST), MOHURD, Wuhan 430074, China
As one of the widely used antibiotics, sulfamethoxazole (SMX) has received increasing attentions on frequent occurrence and high persistence in the aquatic environment [1]. Fenton oxidation process, is a classic and one of the most efficient advanced oxidation processes (AOPs) in treating hazardous and/or hardlydegradable pharmaceuticals and personal care products (PPCPs) as reported in the recent years [2, 3]. Despite that most Fenton technologies can produce ·OH of high oxidation potential (2.8 eV) for rapid and non-selective oxidation of the target recalcitrant organic pollutants, challenges such as acidic reaction circumstances, overload of Fe2+ and low utilization of H2O2 limit their applications in real wastewater treatment [4, 5].
Electro-Fenton (EF) process is a good alternative Fenton process in which H2O2 is generated electrolytically via two electron cathodic reduction of oxygen in acidic medium [6]. In the past decades, a mass of researches have focus on how to raise the onsite cathodic generation efficiency of H2O2, while few studies thought about the simultaneous reduction of Fe3+ to Fe2+ that would also lead to more efficient utilization of H2O2 catalysis as well as lower production of iron sludge [7]. Recently, a method named Fered-Fenton has been developed by adopting cathodic Fe3+ reduction instead of H2O2 generation [8]. Generally, FeredFenton is cost-effective than common EFs, since the applied voltage and current intensity for the reduction of Fe3+/Fe2+ is lower than O2/H2O2 on cathode [9].
Circumstance pH tends to be another factor affecting the application of Fenton technologies in treating actual neutral industrial wastewaters because either Fe2+ or Fe3+ cannot persist in the bulk solution. Several additives such as hydroxylamine (HA) [10, 11], organic chelates [12, 13] could effectively expand the work pH range of Fenton or Fenton-like systems up to neutral since they would maintain essential neutral Fe(Ⅲ)/Fe(Ⅱ) recycles. It has been reported that neutral Fenton-like systems based on iron-ligands have good efficiencies on the degradation of pollutants, but also caused more difficult regeneration of Fe2+ because of the lower redox potential of Fe3+-ligands than free Fe3+ [14]. Furthermore, most related studies used ligands to prompt the in-situ chemical oxidation adopting heterogeneous solid catalysts, e.g., iron oxides, wherein the interfacial surface binding reactions between ligands and metal ion in the solid catalyst have been intensively concerned [15]. In addition, utilization of H2O2 in neutral conditions is questioned since ·OH will be scavenged by increased carbonate [16]. Peroxydisulfate (PDS) is a suitable and favorable alternative powder oxidant since it can be activated to produce sulfate radicals (SO4· -) of higher redox potential (E0 = 2.5–3.1 V) and longer lifetime (3–4 ×10-5 s) as compared to ·OH [17, 18].
Therefore, in this study, a novel neutral Fered-Fenton like/Ox (Fered-FL/Ox) system has been established for more efficient degradation of SMX by using the oxidant PDS, the ligand oxalate and the work cathode pre-anodized Ti@TiO2 electrode. Optimization of operational parameters and exploration of the whole reaction mechanism based on the critical solid-liquid interfacial reactions were proceeded.
All experiments were conducted in a divided threeelectrodes cell reactor with a saturated calomel (SCE) reference electrode, a counter electrode of graphite rod (d = 6 mm), and the working electrode of Ti@TiO2 sheet (20 mm × 20 mm × 0.2 mm) which was pre-anodized with mixed solution of 0.5 mmol/L Fe3+ and 2 mmol/L Ox at a weak voltage for 10 min. 10 mmol/L sodium sulfate (Na2SO4) was chosen as the electrolyte. The Design-Expert 8.0.6 software (Stat-Ease Inc., USA) was used to design the experiments to conduct central composite design (CCD) and to establish response surface methodology RSM models for optimizing four main experiment parameters, i.e., initial pH, cathode voltage, dosage of Ox and Fe2+ at five levels. Upon the optimized parameters and neutral circumstance, attenuated total reflection Flourier transformed infrared spectroscopy (ATR-FTIR) and X-ray photoelectron spectroscopy (XPS) were used to explore the improved heterogeneous-homogenous degradation mechanism based on the Fered-FL/Ox system adopting the Ti@TiO2 electrode.
Degradation of SMX in four comparative systems, i.e., PDS/Ox, Fe2+/Ox-PDS, electro-PDS/Ox (EC-PDS/Ox) and the Fered-FL/Ox, were carried out under the conditions initial sulfamethoxazole (SMX) of 10 mg/L, pH of 6.5 and Ox of 0.4 mmol/L in solution. It was found that the time-dependent mineralization of SMX in all systems could be applied with pseudo first-order reaction kinetic (R2 > 0.95).
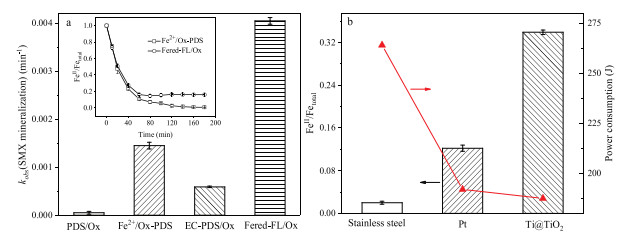
Fig. 1a shows that the Fered-FL/Ox system could achieve significant synergistic mineralization of SMX as compared to the other three systems. Its kobs(SMX mineralization) was 4.1 ×10-3 min-1 which was about 81, 6.9 and 2.8 times larger than the PDS/Ox, EC-PDS/Ox and Fe2+/Ox-PDS systems, respectively. It indicated that Fe2+ could effectively catalyze PDS in producing sulfate radical and oxidizing SMX, while the FeredFL/Ox system would lead to an efficient iron cycle based on the electrochemical reduction of Fe3+-Ox in the neutral reaction circumstance. As presented in the inset of Fig. 1a, the added Fe2+ was rapidly consumed and used up at 60 min in Fe2+/Ox-PDS system, while a stable [FeⅡ]/[Fe]total ratio of 0.16 could be kept in the Fered-FL/Ox system even the reaction proceeded to the reaction time of 180 min. This result indicated that the electroregeneration efficiency of FeⅡ/Ox in the Fered-FL/Ox system could cause continuous catalytic decomposition of PDS and effective mineralization of SMX.

|
Download:
|
| Fig. 1. (a) Comparative kobs(SMX mineralization) at 3 h for PDS/Ox, Fe2+/Ox-PDS, EC-PDS/Ox and Fered-FL/Ox systems. Inset shows the related evolution of FeⅡ/Fetotal in Fe2+/Ox-PDS and Fered-FL/Ox systems. Initial conditions: 10 mg/L SMX, pH 6.5, 0.5 mmol/L PDS, 0.1 mmol/L Fe2+, 0.4 mmol/L Ox, -0.7 V applied voltage and 25 ℃. (b) Evolution of FeⅡ/Fetotal and the related power consumption at 3 h with comparative electrodes. Initial conditions: pH 6.5, 0.1 mmol/L Fe3+, 0.4 mmol/L Ox, -0.7 V applied voltage and 25 ℃). | |
Fig. 1b compares the electro-reduction efficiency of 0.1 mmol/L FeⅢ-Ox at pH of 6.5 using three common metal electrodes/ cathodes, i.e., Ti@TiO2 (20 mm × 20 mm × 0.2 mm), Pt (square flag, 20 mm × 20 mm × 0.2153 mm), and stainless steel (mesh, 20 mm × 20 mm × 0.1897 mm). It can be seen that the Ti@TiO2 electrode achieved a [FeⅡ]/[Fe]total of 0.34, whereas the Pt and stainless steel could only achieve the [FeⅡ]/[Fe]total of 0.12 and 0.02, respectively. Meanwhile, it was interesting to note that the corresponding power consumption of the three electrodes presented a converse behavior. Ti@TiO2 cathode could reduce 0.033 mmol/L Fe(Ⅲ) by consuming only 98 J. It indicated that Ti@TiO2 electrode would be very effective in the electroregeneration of ferric in such iron-based Fenton like systems. It is well known that Pt is inert and commonly have a surface oxide layer due to the spontaneous passivation, while the ATR-FTIR of the commercial stainless steel in adsorbing FeⅢ-Ox exhibited that its surface structure would be more restrict to the solid-liquid interfacial reaction in the Fered-FL/Ox system. Therefore, the significant difference in the three metal electrode might ascribe to the potential surface-binding reactions of FeⅡ/FeⅢ-Ox complexes depending on the specific surface structure which would affect the interfacial electron transfer [18]. More detailed description in the related mechanism will be discussed in the below content.
Thereafter, parameters optimization for the SMX degradation in the Fered-FL/Ox system adopting the Ti@TiO2 cathode was conducted with the four factors and five levels CCD as presented in Table 1. Thirty experimental runs have been designed with the SMX degradation efficiency (kobs) as the response.
|
|
Table 1 The CCD design and response of SMX degradation (kobs) in the Fered-FL/Ox system. |

After the step-wise model fitting by the software, a response surface model for the SMX degradation in the Fered-FL/Ox system could be concluded as expressed by Eq. (1).

|
(1) |
Table 2 variance (ANOVA) of the obtained model. It indicated that this model could be successfully applied for the SMX degradation in the Fered-FL/Ox system. The model value of "prob>F" and F value were < 0.05 and 33.34, respectively. This indicated that both the model terms and the model were significant were significant, there was only a 0.01% chance that a model value this large could occur due to noise. In addition, it should be noted that the "lack of fit P-value" was 0.4712 (>0.1000), suggesting that the lack of fit was not significant and the model had a good predictability. Besides, the value of R2 and "Adeq. precision" reached 0.9532 and 22.57 (>4), respectively, indicating that this model could be used to navigate the design space with an adequate signal and it had a good degree of fitting.
|
|
Table 2 ANOVA (analysis of variance) for the RSM model. |

According to the expression (1), the results of different experimental parameters could be optimized under the remaining experimental conditions: 20 mg/L SMX, 10 mmol/L Na2SO4, 0.5 mmol/L PDS. As shown in Table 3, for achieving "the best degradation efficiency", the optimal parameters were predicted as pH of 5.79, cathode voltage of -0.745 V, Ox dosage of 1.98 mmol/L, Fe2+ dosage of 0.985 mmol/L, by set all related parameters "in range". An estimated value kobs (SMX) of 1.34 × 10-2 min-1 was obtained, which was close to the actual value of 1.44 ± 0.06 × 10-2 min-1 in the Fered-FL/Ox system. Another trial for achieving the target "the most cost-effective" was also calculated by the model, where the parameters were set as "pH equal to 7", "the cathode voltage minimize" (-0.5 V) and "others in range". The model estimated kobs(SMX) was 1.25 × 10-2 min-1 which was also very close to the related actual value of 1.23 ± 0.06 × 10-2 min-1. The above result indicated that the precision of the model was good enough in advising the degradation of pollutants in the Fered-FL/Ox system. In addition, optimal parameters of Ox dosage was 2.45 mmol/L, [Fe2+] dosage was 1.0 mmol/L for the case of "most cost-effective" were noted. The values were slightly higher than the case "best efficiency". It was because that the catalysis activity of FeⅡ-Ox complexes would be lower in the neutral circumstances than the acidic circumstances [19].
|
|
Table 3 The verification for the optimized RSM model. |

{kind=link}
As a result, the related RSM analysis between the initial pH and [Ox] affecting kobs(SMX) was further conducted. It was observed that raising pH would lead to obvious inhibition in the kobs(SMX) since Fe3+ tended to precipitate when [Ox] was too low to effectively maintain the aqueous Fenton like reaction. On the contrary, overloaded free Ox would competitively adsorb on the Ti@TiO2 layer, causing less electrochemical reduction of the soluble Fe (C2O4)33- complex. Therefore, more but appropriate amounts of [Fe2+] and [Ox] would be needed in achieving efficient degradation of SMX in the neutral circumstance as compared to the acidic, since more Fe2+ would be benefit for the activation of PDS but need more Ox to maintain the effective iron cycle in the Fered-FL/Ox system.
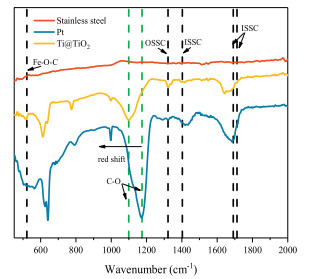
To reveal the interfacial reaction mechanism in the Fered-FL/Ox system depending on the Ti@TiO2 electrode, XPS and ATR-FTIR characterizations were conducted for the three adopted cathodes, which were pre-adsorbed for 10 min in the neutral solution of 0.5 mmol/L Fe3+ and 2 mmol/L Ox under a weak voltage of -0.01 V. Under the neutral conditions, Fe(C2O4)33- would be the dominate FeⅢ-Ox complex and would be absorbed to cathode and then may interact with specific cathode surface to accelerated the process of reduction. From Fig. 2, it can be seen that the obtained Fe 2p spectra of the three electrodes with pre-adsorbed FeⅢ-Ox were rather different. The case of the stainless steel electrode presented an extremely low intensity of Fe 2p indicating difficult adsorption of FeⅢ-Ox on its surface. As for the cases of Pt and Ti@TiO2, the two broad peaks (711.2 and 713.6 eV) and the small shoulder at 709.2 eV could be attributed to the adsorbed FeⅢ-Ox and its electrochemical reduction products FeⅡ-Ox, respectively [19, 20]. This result indicated that FeⅢ-Ox could be effectively adsorbed on the two cathodes surface and reduced. It was interesting to note that the intensity ratio of Fe2+/Fe3+on the Ti@TiO2 electrode increased significantly from 28.2% to 53.5% as compared to the Pt electrode. This indicated that the adsorbed FeⅢ-Ox species would be more easily electrochemically reduced depending on the Ti@TiO2 surface, probably due to TiO2 had more amounts of surface hydroxyl than Pt to combine with FeⅢ-Ox and then easily to start a reduction reaction at a weak voltage. As a result, the electron transportation between FeⅢ-Ox and the cathode could be accelerated.

|
Download:
|
| Fig. 2. The XPS Fe 2p spectra of the stainless steel/Pt/Ti@TiO2 cathodes with preadsorption in the presence of 0.5 mmol/L Fe3+ and 2 mmol/L Ox with a voltage of -0.01 V for 10 min. | |
{kind=link}
The corresponding ATR-FTIR examination was further conducted to verify the critical role of cathode surface hydroxyl in the Fered-FL/Ox system. As shown in Fig. 3, the FTIR patterns of the three electrodes were also different. In general, the formation of FeⅢ-Ox complexes could be roughly divided into two types, one was the outer sphere surface complex (OSSC) and the other was the inner sphere surface complex (ISSC) [21]. In the neutral Fered-FL/Ox system, FeⅢ-Ox complexes would be formed as Fe(C2O4)33- in consistent with the relatively large Langmuir equilibrium constant. From Fig. 3, it can be seen that few peak was found in the case of the stainless steel electrode, verifying this electrode could not adsorb FeⅢ-Ox effectively. While in the cases of Ti@TiO2 and Pt electrodes, the peak at 1394 cm–1 and the peaks at 1710, 1690 cm–1 were clearly observed, which corresponded to the ν (C—O) stretching vibrations involving predominantly the oxygen atom not bonded or bonded to Fe in a 

|
Download:
|
| Fig. 3. ATR-FTIR spectra of the stainless steel/Pt/Ti@TiO2 cathodes with preadsorption in the presence of 0.5 mmol/L Fe3+ and 2 mmol/L Ox with a voltage of - 0.01 V for 10 min. | |
{kind=link}
It was worth noting that an obvious red-shift of the C—O peak at 1109 cm–1 in the case of Ti@TiO2 electrode, suggesting a potential "electron-rich" phenomenon in the bond [27]. This would be attributed to the easier electron transfer through the surface C—O—Ti bond. Therefore, it could be concluded that the surface characteristics of the Ti@TiO2 electrode would be more benefit for the electrochemical reduction of FeⅢ—Ox and lead to the more efficient SMX degradation.
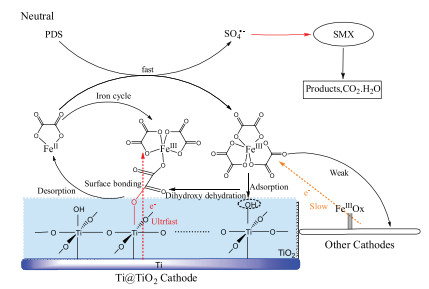
Fig. 4 illustrates the proposed reaction mechanism in the neutral Fered-FL/Ox system depending on the Ti@TiO2 cathode. At first, aqueous neutral Fenton like oxidation of PDS occurred in the presence of FeⅡ—Ox and PDS where SO4· - would be the dominant oxidant [28]. Simultaneously, the efficient electrochemical reduction of the formed FeⅢ—Ox complexes would happen through a series of solid-liquid interfacial reactions. Herein, the main FeⅢ—Ox species Fe(C2O4)33- could be hydrated and electrostatically interacted with the Ti@TiO2 cathode surface through ISSC and form specific C—O—Ti bonds, which would induce ultrafast electron transfer from the cathode to the FeⅢ [29, 30]. It would cause efficient electrochemical generation of FeⅡ-Ox which easily detached to the bulk solution due to its low affinity with the surface hydroxyl groups [30]. The appended FeⅡ species would continuously activate PDS and lead to efficient Fenton like oxidation of SMX.

|
Download:
|
| Fig. 4. Scheme of the proposed reaction mechanisms in the Fered-Fenton like-Ox system. | |
{kind=link}
Therefore, an efficient neutral heterogeneous-homogenous iron cycle would exist in the Fered-FL/Ox system adopting the Ti@TiO2 cathode. It would effectively accelerate the neutral Fenton-like reactions and complete mineralization of SMX with relative low dosage of ferrous catalyst and applied voltage. The result of this study is expected to supply a good alternative in treating complex neutral industrial wastewaters.
AcknowledgmentsThis study is financially supported by the National Natural Science Foundation of China (Nos. 21677055 and 21407052), and the Fundamental Research Funds for the Central Universities, HUST (Nos. 2017KFXKJC004 and 2016YXMS287). Huazhong University of Science & Technology Analytic and Testing Centre is thanked for the advanced analytic operations.
| [1] |
H. Li, D. Cui, J. Liang, H. Cai, Y. Wang, Chin. Chem. Lett. 17 (2016) 1481-1484. |
| [2] |
Y.F. Fang, A.P. Deng, Y.P. Huang, Chin. Chem. Lett. 20 (2009) 1235-1240. DOI:10.1016/j.cclet.2009.05.004 |
| [3] |
J. Anotai, N. Wasukran, N. Boonrattanakij, Chem. Eng. J. 352 (2018) 247-254. DOI:10.1016/j.cej.2018.07.037 |
| [4] |
P.V. Nidheesh, R. Gandhimathi, S.T. Ramesh, Environ. Sci. Pollut. R 20 (2013) 2099-2132. DOI:10.1007/s11356-012-1385-z |
| [5] |
T. Sruthi, R. Gandhimathi, S.T. Ramesh, P.V. Nidheesh, Chemosphere 210 (2018) 38-43. DOI:10.1016/j.chemosphere.2018.06.172 |
| [6] |
P.V. Nidheesh, R. Gandhimathi, Desalination 299 (2012) 1-15. DOI:10.1016/j.desal.2012.05.011 |
| [7] |
P.V. Nidheesh, R. Gandhimathi, N.S. Sanjini, Sep. Purif. Technol. 132 (2014) 568-576. DOI:10.1016/j.seppur.2014.06.009 |
| [8] |
Y. Shih, C. Lin, Y. Huang, Sep. Purif. Technol. 104 (2013) 100-105. DOI:10.1016/j.seppur.2012.11.025 |
| [9] |
A. özcan, Y. ahin, A.S. Koparal, M.A. Oturan, J. Hazard. Mater. 153 (2008) 718-727. DOI:10.1016/j.jhazmat.2007.09.015 |
| [10] |
J. Li, Y. Wan, Y. Li, G. Yao, B. Lai, Appl. Catal. B:Environ. 256 (2019) 117782. DOI:10.1016/j.apcatb.2019.117782 |
| [11] |
L. Chen, J. Ma, X. Li, et al., Environ. Sci. Technol. 45 (2011) 3925-3930. DOI:10.1021/es2002748 |
| [12] |
T. Zhou, T. Lim, Y. Li, X. Lu, F. Wong, Chemosphere 78 (2010) 576-582. DOI:10.1016/j.chemosphere.2009.10.057 |
| [13] |
T. Zhou, X. Zou, X. Wu, J. Mao, J. Wang, Ultrason. Sonochem. 37 (2017) 320-327. DOI:10.1016/j.ultsonch.2017.01.015 |
| [14] |
C.R. Keenan, D.L. Sedlak, Environ. Sci. Technol. 42 (2008) 6936-6941. DOI:10.1021/es801438f |
| [15] |
S.J. Hug, B. Sulzberger, Langmuir 10 (1994) 3587-3597. DOI:10.1021/la00022a036 |
| [16] |
A.G. Trovó, R.F.P. Nogueira, A. Agüera, et al., Water Res. 43 (2009) 3922-3931. DOI:10.1016/j.watres.2009.04.006 |
| [17] |
Y. Aimer, O. Benali, K. Groenen Serrano, Sep. Purif. Technol. 208 (2019) 27-33. DOI:10.1016/j.seppur.2018.05.066 |
| [18] |
Y.Q. Zhang, X.Z. Du, W.L. Huang, Chin. Chem. Lett. 22 (2011) 358-361. DOI:10.1016/j.cclet.2010.10.015 |
| [19] |
H. Xu, Y. Sun, J. Li, F. Li, X. Guan, Environ. Sci. Technol. 50 (2016) 8214-8222. DOI:10.1021/acs.est.6b01763 |
| [20] |
A.P. Grosvenor, B.A. Kobe, M.C. Biesinger, N.S. Mcintyre, Surf. Interface. Anal. 36 (2004) 1564-1574. DOI:10.1002/sia.1984 |
| [21] |
M. Huang, T. Zhou, X. Wu, J. Mao, Water Res. 119 (2017) 47-56. DOI:10.1016/j.watres.2017.03.008 |
| [22] |
P. Persson, K. Axe, Geochim. Cosmochim. Acta 69 (2005) 541-552. DOI:10.1016/j.gca.2004.07.009 |
| [23] |
J. Fujita, A.E. Martell, K. Nakamoto, J. Chem. Phys. 36 (1962) 324-331. DOI:10.1063/1.1732504 |
| [24] |
T. Sun, M. Wang, Surf. Coat. Tech. 205 (2010) 92-101. DOI:10.1016/j.surfcoat.2010.06.019 |
| [25] |
Z. Lin, R. Hu, J. Zhou, et al., Spectrochim. Acta A:Mol. Biomol. Spectrosc. 173 (2017) 527-531. DOI:10.1016/j.saa.2016.09.050 |
| [26] |
K. Axe, M. Vejgarden, P. Persson, J. Colloid Interface Sci. 294 (2006) 31-37. DOI:10.1016/j.jcis.2005.07.013 |
| [27] |
S.J. Gerber, E. Erasmus, Mater. Chem. Phys. 203 (2018) 73-81. DOI:10.1016/j.matchemphys.2017.09.029 |
| [28] |
L. Ling, D. Zhang, J. Fang, C. Fan, C. Shang, Chemosphere 184 (2017) 417-428. DOI:10.1016/j.chemosphere.2017.06.004 |
| [29] |
T.J. Strathmann, A.T. Stone, Environ. Sci. Technol. 36 (2002) 5172-5183. DOI:10.1021/es0205939 |
| [30] |
M.E. Balmer, B. Sulzberger, Environ. Sci. Technol. 33 (1999) 2418-2424. DOI:10.1021/es9808705 |