 2019, Vol. 30
2019, Vol. 30 


b Department of Environmental Science & Engineering, Fudan University, Shanghai 200438, China
Recently, advanced oxidation processes (AOPs) have received attention as powerful processes to treat highly organic loading and less biodegradable wastewater [1]. Hydroxyl radical (·OH), the most widely used reactive species in AOPs with a redox potential of 2.8 eV, is powerful and non-selective for degrading nearly all organic pollutants [2]. Alternatively, sulfate radical (SO4- ·) based AOPs have become a hotspot due to the longer half-life and stronger oxidizing ability of SO4- · (redox potential = 2.5-3.1 eV) relative to ·OH [3].
Sulfate radicals can be produced through homolytic cleavage of persulfate and unsymmetrical cleavage of peroxymonosulfate(PMS) activated by ultraviolet irradiation, heating, and transition metals [4]. However, ultraviolet irradiation or thermal activation needs energy input, while homogenous catalysts of transition metal ions are harmful to organisms, which limited their applications in practical engineering [5]. In recent years, developing transition metal-bearing heterocatalysts has become a strategy to extend the application of PMS activation [5].
Over the past years, spinel-type ferrites with a general formula, MFe2O4 (M = Mn, Fe, Co, Ni, Cu), have been used in lots of applications including high-density magnetic storage, drug delivery, environmental remediation, and catalysis, due to their outstanding properties in optics, electricity and magnetism [6, 7]. Recently, copper ferrite (CuFe2O4) has been demonstrated to be active for PMS activation to degrade organic pollutants [4]. However, the high surface area and unique magnetic properties of MFe2O4 materials lead to their easy aggregation during the practical application, affecting the catalytic efficiency. Therefore, some carbonaceous materials with high electrical conductivity have been employed as the matrix for MFe2O4 to uniformly distribute the active sites and enhance the catalytic performance of MFe2O4 [8].
Among the current carbonaceous support materials, graphene has attracted tremendous attention with the excellent properties of high electrical conductivity, unique mechanical strength, and large specific surface area [6]. Graphite oxide (GO), an important graphene precursor, shares similar sheet structures and properties but with low cost in comparison to graphene [9]. The high sorption capacity and semiconductivity of GO could provide many benefits to their hybrids or composites with metal oxide catalysts [10]. However, to our best knowledge, there is no report on the exploration of CuFe2O4-GO hybrid for activation of PMS to degradation aqueous organic pollutants.
In this study, a new type of CuFe2O4@GO hybrid was synthesized through a sol-gel combustion method and the synthesis parameters were optimized based on activity for activation of PMS to decompose model dye pollutants. The asprepared catalyst was characterized, and its catalytic mechanism of PMS activation was explored. The influences of various experimental conditions on the catalytic degradation process were investigated and explained. The reusability and stability of CuFe2O4@GO were also addressed.
GO was synthesized following Hummers' method with modification [11]. CuFe2O4@GO (mass ratio of 1:1) was prepared using a sol-gel combustion method according to the literature with modification [4]. Typically, Cu(NO3)2·3h2O (1.208 g), Fe (NO3)3·9H2O (4.04 g), and GO (4.2 g) were dissolved in deionized water, referring as solution A. Meanwhile, C6H8O7·H2O (3.1521 g) was added into deionized water to make solution B. Then, solution A was added into solution B drop by drop, and the system then was heated in a water bath to 60 ℃ with stirring continuously for 2 h. Next, the mixture solution was transferred to an oven and dried at 90 ℃ for 7 h until obtain solid composite. After annealing at 300 ℃ for 2 h, the final product was obtained. For a comparison, bare CuFe2O4 was also prepared using the above method without the addition of GO. The synthesis parameters including GO loadings (wt% in raw materials), calcination temperature and time were optimized and determined by catalytic evaluation using methylene blue (MB) as the model organic pollutant.
The as-prepared catalysts were characterized by X-ray diffraction (XRD), scanning electron microscope (SEM), transmission electron microscope (TEM) with energy dispersive X-ray spectrometry (EDS), Fourier transform infrared spectra (FTIR), X-ray photoelectron spectroscopy (XPS), Brunauer-Emmett-Teller (BET) surface area measurement, and electron paramagnetic resonance (EPR). Details information can be found in Text S1 of Supporting information.
Catalytic degradation experiments were conducted in a 250 mL conical flask placed in a thermostat water bath at a room temperature of 25 ℃ with mechanical stirring. After a specified amount of catalyst and model pollutant solution were added with stirring, the reaction was initialized with PMS addition. At given time interval, samples were taken and immediately filtered through a 0.2 μm polytetrafluoroethylene (PTFE) filter membrane into 1.5 mL centrifugal tubes, which contained Na2S2O3 for quenching the reaction. The dye concentration was immediately determined with UV–vis spectrophotometer at different wavelength (MB 664 nm, X-3B 538 nm, AO7 484 nm, and RhB 554 nm).
Effects of various operational parameters on the catalytic degradation reaction were studied including pollutant type, solution pH, catalyst dosage, PMS dosage, initial concentration of organic pollutant, temperature, humic acid (HA), and inorganic anions. The detailed information is shown in Text S2 of Supporting information.
The reusability and stability of CuFe2O4@GO were evaluated and detailed information can be found in of Supporting information.
To comprehensively evaluate the catalytic performance of CuFe2O4@GO and optimize the synthesis parameters, a series of catalytic degradation experiments with model organic pollutant MB were performed using various catalysts prepared under different conditions. The optimal conditions were determined as: GO loading 50%, calcination temperature 300 ℃ and calcination time 2 h, under which CuFe2O4@GO catalyst were prepared and used in the following experiments. The results of optimization are shown in Text S4 and Fig. S1 (Supporting information).
The morphologies of CuFe2O4 and CuFe2O4@GO characterized by SEM and TEM are shown in Fig. 1. As shown in Figs. 1a and c, the CuFe2O4 particles were nanosized with erratic and uneven polygon shapes and agglomerated appearance. The SEM and TEM images of CuFe2O4@GO (Figs. 1b and d) showed that the graphene morphology was clear, and CuFe2O4 nanoparticles were well distributed on the graphene. Undoubtedly, graphene sheets between CuFe2O4 nanoparticles could suppress the agglomeration of CuFe2O4, which was important for the heterogeneous activation process. In addition, hybridizing leaded to decreased magnetic attraction between the particles and reduced the amount of metal in the same quality catalyst.

|
Download:
|
| Fig. 1. SEM images of CuFe2O4 (a) and CuFe2O4@GO (b); TEM images of CuFe2O4 (c) and CuFe2O4@GO (d). | |
{kind=link}
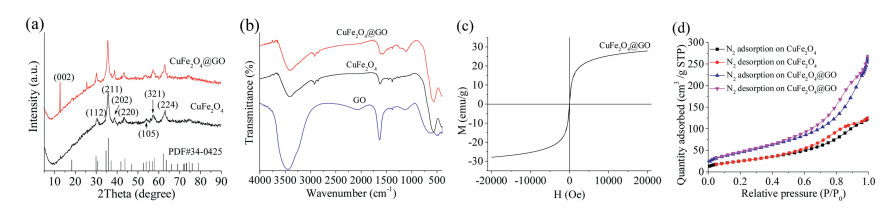
Fig. 2a shows the XRD spectra of the synthesized materials. For the pure CuFe2O4, there were seven intense peaks at 2θ of 30.32°, 35.6°, 38.75°, 43.66°, 54.03°, 57.37° and 63.11° indexed to (112), (211), (202), (220), (105), (321) and (224) planes of copper ferrite, indicating successful formation of the tetragonal CuFe2O4 spinel with considerable degree of crystallization [4]. For CuFe2O4@GO hybrids, all diffraction peaks of CuFe2O4 were detected, and an obvious characteristic diffraction peak of GO appeared at around 12°, indicating that the successful preparation of CuFe2O4@GO catalyst.

|
Download:
|
| Fig. 2. (a) XRD of CuFe2O4 and CuFe2O4@GO; (b) FTIR of GO, CuFe2O4 and CuFe2O4@GO; (c) Magnetization curve of CuFe2O4@GO; (d) N2 adsorption-desorption isotherm of CuFe2O4 and CuFe2O4@GO. | |
{kind=link}
FTIR spectra of GO, CuFe2O4, and CuFe2O4@GO hybrids are showed in Fig. 2b. The peak around 580 cm-1 were assigned as Fe—O bond of tetrahedral geometry [4], verifying the existence of Fe3+ [12]. With the introduction of GO, peaks of C—O—C and O—H stretching vibrations at 1380 cm-1 and 3410 cm-1 emerged in CuFe2O4@GO composites.
The magnetization curve of the prepared CuFe2O4@GO is displayed in Fig. 2c. The saturation magnetization (Ms) value of CuFe2O4@GO was 27.89 emu/g, which is less than that of CuFe2O4 (30 emu/g) [13], which is due to the introduction of non-magnetic GO. However, the CuFe2O4@GO composites dispersed in water still could be easily separated in a few seconds by an external magnet.
Fig. 2d displays the N2 adsorption-desorption isotherms of CuFe2O4 and CuFe2O4@GO. Both samples had the mesoporous structure due to the typical Langmuir-isotherm (type Ⅳ) with a hysteresis loop. As indicated in Table S1 (Supporting information), the specific surface area of CuFe2O4 and CuFe2O4@GO was 92.734 m2/g and 164.507 m2/g, respectively. Large surface area can create more active sites for reaction.
XPS was conducted to investigate the chemical state of the elements in CuFe2O4@GO. The results are summarized in Text S5 and Fig. S2 (Supporting information).
The catalytic degradation of various organic dyes including MB, X-3B, AO7 and RhB was examined, and results are summarized in Fig. 3a. All the dyes could be degraded efficiently within 30 min, and the dye degradability was in the order of X- 3B > AO7 > MB > RhB. Other studies also indicated RhB was a relatively recalcitrant dye pollutant for physiochemical treatments [14]. This results clearly demonstrated that the CuFe2O4@GO was an active catalyst for degradation of a broad spectrum of dye pollutants by PMS oxidation.

|
Download:
|
| Fig. 3. Effects of operational parameters on the catalytic degradation process: (a) dye type, (b) solution pH, (c) catalyst dosage, (d) PMS dosage, (e) dye concentration, (f) temperature, (g) HA and (h) inorganic anions. Typical experimental conditions: MB = 20 mg/L, CuFe2O4@GO = 200 mg/L, PMS = 0.8 mmol/L, T = 25 ℃. | |
{kind=link}
Fig. 3b illustrates the effect of pH on MB catalytic degradation. In the first 5 min of reaction, the efficiency of MB degradation increased from 75.7% to 97.3% when pH was increased from 3 to 11, which may be due to reduction of solvent cage effect by elevated ionic strength [9]. However, after 30 min, the MB degradation rate showed no differences and surpassed 93% at different pH conditions. The addition of PMS reduced the solution pH to 3 without adjustment, and we chose pH 3 for the following studies considering the ease of operation.
The degradation of MB at different catalyst dosages was investigated in this study. As illustrated in Fig. 3c, with increasing CuFe2O4@GO dosage from 100 mg/L to 200 mg/L, the degradation efficiency of MB was improved, which increased from 54.9% to 93.3% within 30 min reaction. This may be attributed to the total surface area of catalyst as well as the active sites increased with increasing dosage, leading to enhanced reaction with PMS to generate more SO4- · and ·OH radicals. However, a further increase of CuFe2O4@GO dosage from 200 mg/L to 400 mg/L showed a very limited elevation on the degradation efficiency of MB. Therefore, the optimal CuFe2O4@GO dosage was 200 mg/L.
Fig. 3d presents the degradation of MB with different PMS dosages. The degradation rate of MB after 30 min was only 69.4% with 0.2 mmol/L of PMS, while the degradation rate value climbed to 93.3% at the PMS dosage of 0.8 mmol/L. Consequently, with the increase of PMS dosage, more radicals were expected to be produced to decompose MB. Whereas, when the PMS dosage further increased to 1 mmol/L, the degradation efficiency of MB showed a slight decline. As reported in the literature, the excessive PMS would play a negative role for the radicals-facilitated degradation of organic pollutants due to the reaction between SO4- · and ·OH (Eq. 1) as well as their recombination (Eqs. 2 and 3), and the PMS scavenging (Eqs. 4 and 5) [15, 16]. Therefore, the optimal PMS dosage was 0.8 mmol/L.

|
(1) |

|
(2) |

|
(3) |

|
(4) |

|
(5) |
Fig. 3e presents the effect of initial MB concentration. With increasing initial pollutant concentration from 10 mg/L to 40 mg/L, the degradation efficiency in 30 min decreased substantially from 96.0% to 57.5%. This may be due to that the large amount of MB molecules would occupy the active sites on catalyst surface, while fixed dosage of PMS is not sufficient for the complete elimination of the total MB [17]. In addition, insufficient amounts of radicals might result in the competition between the MB parent molecules and the intermediates for radicals.
Fig. 3f illustrates the temperature effect on the catalytic degradation process. In the first 5 min of the reaction, the degradation efficiency of MB significantly increased from 52.2% to 87.3% with increasing the temperature from 5 to 45 ℃. However, the final degradation efficiency of MB after 30 min at all temperatures exceeded 91% without any remarkable disparity. Therefore, we chose the mild reaction temperature of 25 ℃ for the catalytic degradation experiments. In addition, the activation energy (Ea) of the reaction on the catalyst surface of CuFe2O4@GO was based on the Arrhenius equation and was determined as 30.0 kJ/mol, which is comparable with those of MnFe2O4 and MnFe2O4-graphene catalyst [6].
Natural organic matter (NOM) is usually considered as a radical scavenger via competing for SO4- ·/·OH radicals, thereby reducing the degradation of organic pollutants by AOPs. This study used HA as the surrogate of NOM to explore the effect of NOM on the catalytic degradation process. With increasing the HA concentration from 0 to 20 mg/L, the degradation efficiency of MB decreased from 93.3% to 81.1% within 30 min (Fig. 3g), which can be attributed to the competition of HA with MB for generated radicals and active sites on CuFe 2O4@GO catalyst [18].
The inorganic anions such as Cl-, SO42-, HCO3-, NO3-, and H2PO4- are commonly existing in wastewater, and thereby their effects on the catalytic degradation process were investigated. As depicted in Fig. 3h, Many anions such as Cl-, SO42- and especially H2PO4- showed negative effects on the MB degradation. These inorganic anions introduced into the solution would compete with MB molecules for limited active sites on CuFe2O4@GO. Moreover, inorganic anions are generally considered as scavengers of radicals (e.g., HSO5-, SO4- · and ·OH) [19]. However, HCO3- exhibited a slightly positive effect on the degradation of MB, which may be related with its promoting effect on stimulating PMS decomposition into SO4- · and ·OH [20]. The positive effects of NO3- may be due to its promoting effect on the production of active oxygen [21].
As shown in Fig. S3 (Supporting information), the degradation efficiency of MB by CuFe2O4@GO/PMS showed only a slight decline from 93.3% to 81.1% after five cycles of experiments, indicating a promising reusing performance of CuFe2O4@GO for practical application. During the recycling experiments, the concentrations of leached Cu and Fe from CuFe2O4@GO was low; only 0.26 mg/L Cu and 0.15 mg/L Fe were detected in the first cycle, and then dropped to 0.07 mg/L and 0.02 mg/L in the fifth cycle.
To identify the contribution of homogeneous activation of PMS by metal ions released from CuFe2O4@GO, 0.3 mg/L of Cu2+ and Fe3+ were added to the solution to trigger the catalytic degradation of MB by PMS oxidation. The degradation efficiency of MB was improved less than 10% by either Cu2+ or Fe3+ (Fig. S4 in Supporting information), indicating the leached metal ions from CuFe2O4@GO made a very limited contribution for the activation of PMS, and the heterogeneous catalysis played the dominant role.
According to the literature, both SO4- · and ·OH radicals can be generated during the activation of PMS by the metal-based catalysts [22]. To identify the possible roles of radicals in MB degradation in the CuFe2O4@GO/PMS system, EtOH and TBA, radical scavengers, were added into the system to capture radicals. EtOH can capture SO4- · and ·OH, while TBA can capture ·OH. With the addition of 0.25 mol/L TBA and EtOH, the degradation efficiency of MB was declined to 87.6% and 74.1%, respectively; increasing TBA and EtOH concentration to 0.5 mol/L resulted in further decrease of degradation efficiency to 67.9% and 41.5%, respectively (Fig. 4a). The much higher inhibitory effect of EtOH in comparison to TBA suggests that both ·OH and SO4- ·radicals played the important role during the activation of PMS by CuFe2O4@GO.

|
Download:
|
| Fig. 4. (a) Effect of scavengers (EtOH and TBA) on the degradation of MB by CuFe2O4@GO/PMS; (b) EPR spectra of DMPO-OH and DMPO-SO4 adducts in the activation of PMS (●: DMPO-OH, ▼: DMPO-SO4). Experimental conditions: MB = 20 mg/L, catalyst = 200 mg/L, PMS = 0.8 mmol/L, T = 25 ℃, DMPO = 5 mmol/L | |
{kind=link}
Furtherly, the EPR was used to detect the active radicals using DMPO as the spin trapping agent in the CuFe2O4@GO/PMS system. As displayed in Fig. 4b, four weak signals of DMPO-OH adduct (intensity ratio = 1:2:2:1) was formed in PMS solution in the absence of catalyst, which could be attributed to the fission of S-O bond in PMS to form hydroperoxide (HO2-) (Eq. 6) [23].

|
(6) |
For the CuFe2O4/PMS and CuFe2O4@GO/PMS system, signals of both DMPO-OH and DMPO-SO4 were observed [24], agreeing with the radical quenching study (Fig. 4a). The intensity of DMPO-OH and DMPO-SO4 in CuFe2O4@GO/PMS system was higher than CuFe2O4/PMS system, indicating an enhanced activation of PMS by CuFe2O4@GO.
The XPS spectrum of used CuFe2O4@GO was investigated to explore the roles of Fe and Cu in PMS activation. As reveled in Fig. S5a (Supporting information), the shoulder peak of Cu(Ⅱ) at 934.4 eV completely disappeared and the area of peak at 933.7 eV became larger, which could be ascribed to the transformation of Cu(Ⅱ) to Cu(Ⅰ) in Cu 2p3/2 after activation [25]. However, a new peak at 936.5 eV appeared, which is assigned to Cu(Ⅱ) according to the literature [26]. Therefore, during the activation process, Cu(Ⅱ)/Cu(Ⅰ) redox couple may play the dominant role in PMS activation. The XPS spectra of Fe 2p after oxidation reaction had no obvious change (Fig. S5b in Supporting information), which suggests an abstinent role of Fe in the activation process [27]. Based on the above experimental results, the catalytic mechanism of CuFe2O4@GO for PMS activation was proposed as follows (Eqs. 7- 14):

|
(7) |

|
(8) |

|
(9) |

|
(10) |

|
(11) |

|
(12) |

|
(13) |

|
(14) |
In summary, a promising CuFe2O4@GO catalyst for PMS activation was synthesized, which had the advantages of high catalytic efficiency, easy recoverability, large surface area and evenly distribution of active sites, and high reusability and stability. The mechanistic study indicated the Cu(Ⅱ)/Cu(Ⅰ) redox couple on CuFe2O4@GO played the dominant role in catalytic process to active PMS to produce SO4- ·and ·OH radicals, which proceeded the oxidative degradation of organic pollutants. Many water chemistry parameters could significantly influence the catalytic degradation process. Increasing the solution pH and temperature showed a positive effect on the PMS activation. The NOM and most inorganic ions (e.g., Cl-, SO42- and H2PO4-) would inhibit the catalytic degradation of organic pollutants due to competition for limited active sites on CuFe2O4@GO and the generated reactive radical species.
AcknowledgmentsThe authors thank the Collaborative Innovation Plan of Hubei Province for Key Technologies in the Eco-Ramie Industry. This work was financially supported by the Natural Science Foundation of Hubei Province, China (No. 2018CFB515). Dr. Jie Fu thanks the financial support from the National Natural Science Foundation of China (No. 41701541).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.05.039.
| [1] |
J. Fu, G.Z. Kyzas, Chin. J. Catal 35 (2014) 1-7. DOI:10.1016/S1872-2067(12)60724-4 |
| [2] |
X. Yang, X. Cheng, A.A. Elzatahry, et al., Chin. Chem. Lett. 30 (2019) 324-330. DOI:10.1016/j.cclet.2018.06.026 |
| [3] |
T. Olmez-Hanci, I. Arslan-Alaton, Chem. Eng. J. 224 (2013) 10-16. DOI:10.1016/j.cej.2012.11.007 |
| [4] |
Y. Ding, L. Zhu, N. Wang, H. Tang, Appl. Catal. B 129 (2013) 153-162. DOI:10.1016/j.apcatb.2012.09.015 |
| [5] |
S. Luo, L. Duan, B. Sun, et al., Appl. Catal. B 164 (2015) 92-99. DOI:10.1016/j.apcatb.2014.09.008 |
| [6] |
Y. Yao, Y. Cai, F. Lu, et al., J. Hazard. Mater 270 (2014) 61-70. DOI:10.1016/j.jhazmat.2014.01.027 |
| [7] |
K. Wang, P. Yang, R. Guo, X. Yao, W. Yang, Chin. Chem. Lett. (2019), doi: http://dx.doi.org/10.1016/j.cclet.2019.04.005.
|
| [8] |
J. Zhu, J. He, ACS Appl. Mater. Inter. 4 (2012) 1770-1776. DOI:10.1021/am3000165 |
| [9] |
J. Fu, G.Z. Kyzas, Z. Cai, et al., Chem. Eng. J. 335 (2018) 290-300. DOI:10.1016/j.cej.2017.10.163 |
| [10] |
Z. Cai, A.D. Dwivedi, W.N. Lee, et al., Environ. Sci.-Nano 5 (2018) 27-47. DOI:10.1039/C7EN00644F |
| [11] |
W.S. Hummers, R.E. Offeman, J. Am. Chem. Soc. 80 (1958) 1339-1339. DOI:10.1021/ja01539a017 |
| [12] |
F. Davar, M. Salavati-Niasari, J. Alloy. Compd. 509 (2011) 2487-2492. DOI:10.1016/j.jallcom.2010.11.058 |
| [13] |
X. Zhang, M. Feng, R. Qu, et al., Chem. Eng. J. 301 (2016) 1-11. DOI:10.1016/j.cej.2016.04.096 |
| [14] |
J. Wang, Y. Lv, L. Zhang, et al., Ultrason. Sonochem. 17 (2010) 642-648. DOI:10.1016/j.ultsonch.2009.12.016 |
| [15] |
X. Wang, L. Wang, J. Li, et al., Sep. Purif. Technol. 122 (2014) 41-46. DOI:10.1016/j.seppur.2013.10.037 |
| [16] |
H. Lin, J. Wu, H. Zhang, Chem. Eng. J. 244 (2014) 514-521. DOI:10.1016/j.cej.2014.01.099 |
| [17] |
T. Zeng, M. Yu, H. Zhang, et al., Sci. Total. Environ. 593- 594 (2017) 286-296. |
| [18] |
X. Duan, Z. Ao, L. Zhou, et al., Appl. Catal. B 188 (2016) 98-105. DOI:10.1016/j.apcatb.2016.01.059 |
| [19] |
X. Lei, X. Li, Z. Ruan, et al., J. Mol. Liq. 266 (2018) 122-131. DOI:10.1016/j.molliq.2018.06.053 |
| [20] |
L. Zhao, Z. Sun, J. Ma, H. Liu, J. Mol. Catal. A 322 (2010) 26-32. DOI:10.1016/j.molcata.2010.02.007 |
| [21] |
Y.H. Huang, Y.F. Huang, C.I. Huang, C.Y. Chen, J. Hazard. Mater 170 (2009) 1110-1118. DOI:10.1016/j.jhazmat.2009.05.091 |
| [22] |
J.E. Bennett, B.C. Gilbert, J.K. Stell, J. Chem. Soc. Perkin Trans. 2 (1991) 1105-1110. |
| [23] |
O.S. Furman, A.L. Teel, R.J. Watts, Environ. Sci. Technol. 44 (2010) 6423-6428. DOI:10.1021/es1013714 |
| [24] |
G.D. Fang, D.D. Dionysiou, S.R. Al-Abed, D.M. Zhou, Appl. Catal. B 129 (2013) 325-332. DOI:10.1016/j.apcatb.2012.09.042 |
| [25] |
Y. Xu, J. Ai, H. Zhang, J. Hazard. Mater 309 (2016) 87-96. DOI:10.1016/j.jhazmat.2016.01.023 |
| [26] |
Y. Wang, H. Zhao, M. Li, J. Fan, G. Zhao, Appl. Catal. B 147 (2014) 534-545. DOI:10.1016/j.apcatb.2013.09.017 |
| [27] |
L. Chen, D. Ding, C. Liu, et al., Chem. Eng. J. 334 (2018) 273-284. DOI:10.1016/j.cej.2017.10.040 |