 2019, Vol. 30
2019, Vol. 30 


In recent years, the excessive wastewater discharge containing organic and inorganic pollution from different industry like textiles, printing, leather and cosmetics, is endangering the environment and ecology, and reversely limited the industry development [1-4]. To deal with the problem, some strategies were adopted usually, such as membrane technology [5, 6], biodegradation [7, 8] and physicochemical processes [9-11]. So far, there is no single method is as effective as the method we mentioned above to reasonably handle wastewater composed of mixture toxic chemicals (phenols, dyes, pesticides, organic solvents, etc.). Up to now, advanced oxidation processes (AOPs) are chosen as an effective solution with gentle operation condition of room temperature and pressure. Different from the conventional wastewater remediation methods, the in situ generated hydroxyl radicals (·OH) counts in the AOPs strategy, which owns a relatively strong oxidation potential of 2.80 V [12, 13]. Fenton process is used as a kind of conventional AOPs, where the hydrogen peroxide (H2O2) plays as the oxidant and meanwhile the Fe2+ ion plays as the catalyst, involving a redox reaction along with releasing enormous amount of ·OH in a while [14-16]. And the main active species generation mechanism in the Fenton process can be divided and reduced into three steps, as shown in Eqs. (1–3).

|
(1) |

|
(2) |

|
(3) |
According to Eq. (2), it can be known that the mainly efficiency limitation in the Fe(Ⅱ)/H2O2 system is the comparatively low constant of k2 corresponding to the step of Fe3+/Fe2+ cycle. Besides, it is also is confronted with some trouble in practical application, for instance, the high cost, the limited pH range, the sustainably produced iron sludge, and challenges in reusing the homogeneous catalyst (Fe2+). Even worse, a high concentration of Fe3+ ions would lead to a reaction poisoning in conventional Fenton process. Thus, it does call for a valid solution to accelerate the reaction of the ratelimiting step of Eq. (2) to promote the Fe3+/Fe2+ cycle process. On one hand, it can help produce more Fe2+ ions. On the other hand, it can also reduce the generation of iron sludge.
Based on our previous work, it was found that metal sulfides (MoS2, WS2, CoS2, etc.) could greatly promote Fenton reaction as cocatalysts, because of the exposed reductive metal sites on the surface of cocatalysts [4, 17]. However, it makes a potential secondary contamination risk to the environment for the release of hydrogen sulphide. Herein, we employed MoO2 instead of MoS2 as a cocatalyst to increase the H2O2 decomposition efficiency and decrease the consumption of H2O2 and Fe2+ in AOPs for the remediation of Lissamine rhodamine B (L-RhB). Generally, ·OH radicals are considered as the main reactive oxygen species (ROS) for oxidation of organic pollutants in Fenton Process. Interestingly, in the MoO2 co-catalytic system, it is found that the singlet oxygen (1O2) acts as the main ROS for the oxidation, which is demonstrated to be involved with the transformation from ·OH radicals to singlet oxygen (1O2) on the surface of MoO2. Meanwhile, it can also achieve the goal of reducing of Cr(Ⅵ) in suspension, which shows a synchronous REDOX activity over Fenton reaction.
First of all, to optimize the reaction conditions, the effects of pH value, Fe2+ concentration, hydroxyl peroxide (H2O2) and MoO2 powder dosage in the degradation of L-RhB were investigated in Fig. 1. In our case, all the Fenton experiments were carried out under dark situation at room temperature. As a result, the optimum condition of MoO2 cocatalytic Fenton reaction for the remediation of L-RhB is fixed at: pH ~3.4, FeSO4·7H2O dosage of 30 mg/L, H2O2 concentration of 0.4 mmol/L and MoO2 powder dosage of 30 mg/L. As can be seen in Fig. 1a, when pH value is near neutral condition, the efficiency was obviously inhibited, which is ascribed to the loss of dissolution of iron ions. Additionally, in Fig. 1b, the activity of degrading L-RhB was not significantly improved with the continuous increase of Fe2+ concentration, which indicates that H2O2 has achieved a complete decomposition and the excessive iron ions cannot significantly produce more ROS. Moreover, when H2O2 added slowly, the Fenton efficacy firstly shows an increasing trend, and then followed by a downward trend because that H2O2 possesses both oxidative and reductive ability (Fig. 1c). Seen from Fig. 1d, as the adding amount of MoO2 increased, the Fenton activity tends to increase, indicating that the cocatalyst plays a vital role on reaction kinetics of the Fenton reaction. In addition, we have carried out the apparent kinetic calculation of MoO2 cocatalytic Fenton reaction for the remediation of L-RhB, as shown in Fig. S1 (Supporting information). As a result, according to the apparent kinetic equation (Eq. 4), the total reaction constant of "a + b+c" equals to 2.229, which implies that the adding of FeSO4·7H2O, H2O2 and MoO2 are beneficial to improving the efficiency of Fenton process.

|
(4) |

|
Download:
|
| Fig. 1. Fenton degradation performance of L-RhB (20 mg/L) by adding cocatalyst of MoO2 at different conditions: (a) different pH value; (b) different FeSO4·7H2O concentration; (c) different H2O2 dosage; (d) different MoO2 concentration. (e) Degradation of L-RhB (20 mg/L) over different Fenton systems. (f) Cycling test of the MoO2 cocatalytic Fenton reaction for the degradation of L-RhB (20 mg/L). (g) XRD patterns of co-catalyst of MoO2 before and after cycle test. (h) High performance liquid chromatography (HPLC) spectra of different reaction products for the L-RhB (20 mg/L) degradation over different Fenton systems. | |
{kind=link}
where V is the reaction rate of the Fenton degradation reaction; c is the concentration of Lissamine rhodamine B; t is time.
Based on the above experimental results, all the Fenton systems for the degradation of L-RhB are fixed under the optimal reaction condition: pH ~3.4, FeSO4·7H2O dosage of 30 mg/L, H2O2 concentration of 0.4 mmol/L and MoO2 powder dosage of 30 mg/L, as shown in Fig. 1e. The MoO2+FeSO4+H2O2 system can degrade 99% of L-RhB within 3 min, whereas the conventional Fenton system merely degraded 60% in 3 min. And it was found that the MoO2 Fenton system can develop the TOC removal rate of L-RhB from 30.53% to 70.53% comparing to the conventional Fenton (Fig. S2 in Supporting information). In addition, although MoO2 is a kind of semiconductor, it has no photocatalytic activity for degrading L-RhB under the simulated solar light irradiation (Fig. S3 in Supporting information). Meanwhile, in the absence of Fe2+ ions or H2O2, the corresponding efficiencies for the remediation of L-RhB could be neglected (< 10%). Various concentrations of L-RhB were also be degraded by the conventional Fenton and MoO2 cocatalytic Fenton systems (Fig. S4 in Supporting information). When the concentration of L-RhB increases from 20 mg/L to 80 mg/L, it shows a downward trend. Furthermore, it has been explored the intermediates by high performance liquid chromatography (HPLC) during the Fenton process (Fig. 1h). Three systems (blank LRhB, conventional Fenton, MoO2 cocatalytic Fenton) were compared for HPLC detection. It can be clearly elucidated that the peak of residual L-RhB corresponding to the MoO2 cocatalytic system is the weakest, and the peaks of intermediate products are also the least. In addition, the cyclic experiment of cocatalytic Fenton system is conducted as well, as shown in Fig. 1f and g. The degradation rate can keep up to 99.9% after 5 cycles, and the mass and microstructure of MoO2 are almost unchanged after cycle test (Fig. S5 in Supporting information), further confirming the stability of MoO2 during the Fenton reaction. The low dosage of Fe2+ (0.105 mmol/L) and H2O2 (0.4 mmol/L) used in the MoO2 cocatalytic Fenton reaction can effectively prevent the formation of iron sludges, thus avoiding catalyst poisoning. To further understand the decomposition mechanism of organic pollutant, the mass spectra of intermediates in LC chromatogram during the degradation process of L-RhB over MoO2 cocatalytic Fenton system was analyzed, as shown in Fig. S6 (Supporting information). The whole degradation process has gone through deoxygenation, debenzene ring, deethylation and dehydroxylation steps. As a result, the L-RhB was finally decomposed into small fatty molecule of CH3COOH.
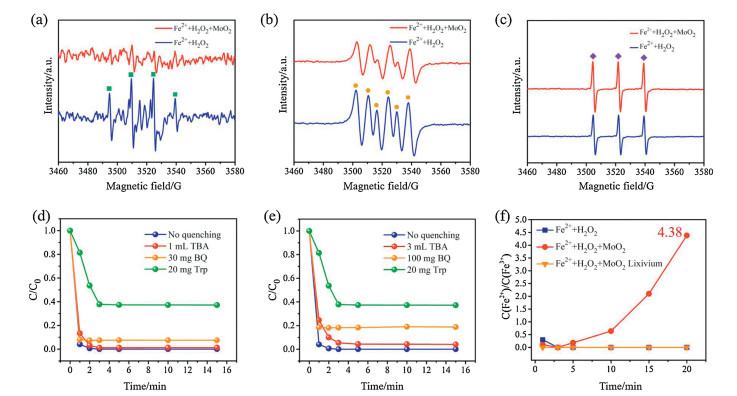
To study the mechanism of MoO2 cocatalytic Fenton process, it is of great significance to identify the ROS during the reaction. It has been reported that ·OH, as well as ·O2- and 1O2 co-work in Fenton process [18]. Moreover, some researchers have reported that the 1O2 can be generated by the disproportionation of ·OH radicals [19]. In this case, 5, 5-dimethyl-1-pyrrolidine-N-oxide (DMPO) and 2, 2, 6, 6-tetramethylpiperidine (TEMP) are employed to capture the ·OH/·O2- and 1O2, respectively, for the EPR characterization (Figs. 2a-c). It can be clearly seen that the MoO2 cocatalytic Fenton system exhibits an obvious decrease of ·OH/·O2- radicals, whereas a slight increase of ·O2- radicals, compared with that of conventional Fenton reaction. This result is totally opposed to previous research [20], which means that the MoO2 cocatalytic Fenton system undergoes different reaction paths. The decrease generation of ·OH and ·O2- radicals may contribute to the increased 1O2 generated in MoO2 cocatalytic system. To further investigate the mechanism of MoO2 cocatalytic Fenton process, some quenching tests have been conducted under various conditions. Tert-butyl alcohol (TBA), p-benzoquinone (BQ) and tryptophan (Trp) were used as the quenching agents of ·OH, ·O2- and 1O2, respectively [21]. In Fig. 2d, it is found that the adding of TBA and BQ cannot effectively inhibit the reaction; however, when Trp was added, the Fenton efficiency was obviously constrained. To be more specific, ~3-fold amounts of the TBA and BQ are added in the reaction, and it merely showed a slight inhibitory effect (Fig. 2e). Based on these results, we can conclude that 1O2 radicals play a vital role in cocatalytic Fenton reaction. Additionally, the concentration of Fe2+ ion in Fenton is an important quantitative standard. Seen from Fig. 2f, the Fe2+/Fe3+ ratio increased to 4.38 in MoO2 cocatalytic system compared with conventional Fenton reaction, indicating the enhancing conversion efficiency from Fe3+ to Fe2+ by the cocatalytic effect of MoO2. In addition, it has been added the data of Fenton degradation performance of L-RhB by adding Fe3+ instead of Fe2+. Seen from Fig. S7 (Supporting information), the results showed that the activity of Fe3++H2O2 was very poor for the degradation of L-RhB, but once MoO2 was added, the Fenton activity of Fe3+ decomposition of H2O2 was greatly increased, which indicated that MoO2 could rapidly reduce Fe3+ to Fe2+, thus showing excellent Fenton activity.

|
Download:
|
| Fig. 2. (a) EPR spectra for the detection of ·OH (the EPR signals are marked as green square) in the presence of DMPO by using water as the solvent. (b) EPR spectra for the detection of ·O2- (the EPR signals are marked as yellow circle) in the presence DMPO by using methanol as the solvent. (c) EPR spectra for the detection of 1O2 (the EPR signals are marked as purple rhombus) in the presence of 2, 2, 6, 6-tetramethylpiperidine (TEMP) by using water as the solvent. (d) Degradation curve of L-RhB (20 mg/L) over cocatalytic Fenton system in the presence of different radical scavengers including BQ and Trp for the quenching of ·OH, ·O2- and 1O2, respectively, under the optimal situation. (e) Increasing TAB and BQ for the quenching of ·OH and ·O2-, respectively, for the degradation of L-RhB (20 mg/L). (f) Fe2+/Fe3+ ratio during the Fenton reaction (blue line: conventional Fenton process, red line: MoO2 cocatalytic Fenton process). | |
{kind=link}
Although it has been proved that the MoO2 can promote the conversion from Fe3+ to Fe2+ during the Fenton reaction, it cannot determine whether the reduction process occurs in solution or on the surface of MoO2. Thus, the coumarin and benzoic acid are employed as the trapping agents for the trapping of ROS in the solution and on the surface of MoO2, respectively. Coumarin has a weak polarity, and it is not easily soluble in water, thus cannot adsorb on the surface of MoO2 to capture ·OH. However, benzoic acid is negatively charged in aqueous solution and easily adsorbed on MoO2 (When pH value is 3.4, the MoO2 surface is positively charged, as shown in Fig. S8 in Supporting information). It can be seen that the "Coumarin+H2O2+Fe2+ +MoO2" shows an obvious decrease signal for the trapping of ·OH in the solution, compared with that of "Coumarin+H2O2+Fe2+ " (Fig. 3a). On the contrary, the "Fe2+ +H2O2+MoO2+C6H5COOH" shows a significant increase signal for the trapping of ·OH, compared with that of "Fe2+ +H2O2+C6H5- COOH" (Fig. 3b). Hence, we can conclude that the cocatalytic reaction mainly occurs on the surface of MoO2. Although ·OH radicals are detected on the surface of MoO2 in the cocatalytic Fenton system, it does not mean that ·OH play a direct role in the oxidation reaction. Since ·OH radicals have a relatively short lifetime (< 2 ns), it is reasonable to believe that ·OH radicals are the main precursors for the formation of 1O2. 1O2 has a relatively long lifetime (2~3.5 μs) and can be diffused into aqueous solution to oxidize and degrade the organic pollutants. It is also worth mentioning that we cannot rule out that ·O2- can also be converted to 1O2 in the presence of Mo6+ [21].

|
Download:
|
| Fig. 3. (a) Coumarin and (b) benzoic acid fluorescence spectrophotometry for the detection of ·OH; (c) Mo 3d XPS spectrum of MoO2; (d) Fe 2p XPS spectrum of iron adsorbed on MoO2 surface; (e) Schematic illustration of the cocatalytic mechanism of MoO2 in Fenton reaction; (f) Molybdenum ions (Mox+) cocatalytic Fenton reaction for the degradation of L-RhB (20 mg/L) (MoO2 was immersed in the solution for different times (pH 3.6): 0 min, 30 min, 120 min); (g) Cycle test of Mox+ cocatalytic Fenton system for the remediation of L-RhB (20 mg/L) (adding 4 μL of H2O2 for each cycle); (h) MoO2 cocatalytic Fenton reaction for the reduction of Cr(Ⅵ) (15 mg/L, K2Cr2O7); (i) Various control experiments for the reduction of Cr(Ⅵ) (15 mg/L, K2Cr2O7). | |
{kind=link}
Since it has been known that the cocatalytic reactions occur mainly on the surface of MoO2, studying the surface chemistry of the catalyst is crucial to reveal the cocatalytic Fenton mechanism. Mo 3d XPS spectra were employed to study the surface chemical environment of MoO2 powder, as shown in Fig. 3c. As to the MoO2 powder, there are two types of molybdenum ions on the surface: Mo4+ and Mo6+. Seen from Fig. 3d, it is confirmed the existence of Fe3+ ions on the catalysts surface, whose characteristic binding energy located at 710.5 eV [22]. This founding suggests the occurring of Fe3+/Fe2+ cycle reaction on the surface of MoO2. To further investigate the reaction mechanism of MoO2 cocatalytic Fenton reaction, the effect of MoO2 powder on the Fe3+/Fe2+ conversion was studied in this case. The concentration of ferrous and ferric ions in the solution can be observed by using UV–vis analysis with potassium thiocyanate and 1, 10-ophenanthroline acting as the probes for the detection of Fe3+ and Fe2+, respectively (Fig. S9 in Supporting information). It can be observed that Fe2+ was instantly oxidized to Fe3+ by H2O2 (Eq. 1). Interestingly, the addition of MoO2 powder can significantly increase the Fe2+ ions but decrease the Fe3+ ions in solution (Eq. 5 and Fig. S9). It can be concluded that the exposed metallic active sites of Mo4+ with reductive properties over MoO2 powder are expected to accelerate the rate-limiting step of Fe3+/Fe2+ conversion and promote the decomposition of H2O2 to form ·O2- and ·OH radicals. Moreover, the H2O2 decomposition efficiency can also be obviously increased by the adding of MoO2 powder in the Fenton reaction (Fig. S10 in Supporting information). Based on above analysis, it can be proposed the possible mechanism, as shown in Fig. 3e. The first step was involved with the exposed Mo4+ for the reduction of Fe3+ adsorbed on the MoO2 surface, which could accelerate the ratelimiting step of Fe3+/Fe2+ conversion (Eq. 5). Simultaneously, the Fe2+ ions in solution reacted with O2 to produce ·O2- radicals (Eq. 6). Subsequently, the H2O2 reacted with ·O2- or Fe2+ to produce ·OH radicals (Eqs. 7 and 8). Due to the short lifespan of ·OH radicals, the generated ·OH radicals would transform into 1O2 on the MoO2 surface by the disproportionation reaction (Eq. 9). As a result, the enhanced efficiency of Fe3+/Fe2+ conversion would decrease the Fe3+ amount and weaken the catalysts poisoning during the Fenton reaction. And the generated 1O2 would migrate into solution to degrade the organic pollutants.

|
(5) |

|
(6) |

|
(7) |

|
(8) |

|
(9) |
In our case, MoO2 powder is ineluctably dissolved in acidic aqueous solution. Therefore, it is important to study the effect of dissolved molybdenum ions (Mox+) on Fenton reaction. We soak the MoO2 powders in an acidic water solution (pH 3.6) to ensure adequate molybdenum ions dissolving out. It has monitored the total content of Mox+ in the solution at different time by ICP test, which shows that the concentration of dissolved Mox+ would gradually reach a maximum value of 0.61 mg/L at about 90 min, and it would not to continue to increase (Fig. S11 in Supporting information). This shows that molybdenum ions do not dissolve continuously, so the possibility of secondary pollution can be excluded. The MoO2 was immersed in the solution for 0 min, 30 min and 120 min, and the supernatant is used for the Fenton reaction, as shown in Fig. 3f. It is found that with the extension of immersing time, the Fenton activity for the degradation of L-RhB increased first and then decreased owing to the that excessive dissolution of Mox+ ions can also consume the ROS. Different from the stable MoO2 cocatalytic Fenton system (Fig. 1f), the Mox+ cocatalytic Fenton reaction shows a poor stability for the degradation of L-RhB (Fig. 3g). It can be found that after two cycles, the Fenton efficiency has been reduced to 50%. And after three cycles, the efficiency merely achieved 30%. Overall, we can conclude thatMox+ ions in solution could have an impact in Fenton process, but not so competitive with the Mox+ ions on MoO2's surface.
Since it has been proved that molybdenum ions on the surface of MoO2 can promote the conversion from Fe3+ to Fe2+, we cannot ignore the reduction performance of MoO2 in the Fenton reaction. It is well known that inorganic contamination is a significant environmental hazard to drinking water treatment. To be specific, hexavalent chromium species Cr(Ⅵ), are highly toxic agents, acting as carcinogens, mutagens, and teratogens in biological systems [23]. According to the U.S. EPA action level, the standard for chromium is 0.1 mg/L for Cr [24]. Commonly, hexavalent chromium exists in waste water as oxyanions, such as chromate(CrO42-)and dichromate (Cr2O72-), which do not precipitate easily using conventional precipitation methods. Thus, adsorption technology, as one of the most promising techniques for removal of chromium from industrial wastewaters, has been employed for many years [25, 26]. However, the adsorption method cannot only remove Cr(Ⅵ) but also recover them back into the industrial process [27]. Thus, it is imperative to develop an effective method to restrain the Cr(Ⅵ) back to the industrial process. Chemical redox followed by precipitation is often employed for the remediation of Cr(Ⅵ) [28-30]. In our previous work, it was found that Fenton system could reduce Cr(Ⅵ) to Cr(Ⅲ) [1], via the cocatalytic effect of WS2. However, sulfides can release hydrogen sulfide and cause the potential risk of secondary pollution. In this work, MoO2 was employed as cocatalyst in Fenton process for the reduction of Cr(Ⅵ), owing to the exposed metallic Mo4+ sites. The Cr(Ⅵ) reduction rate can be enhanced about 40% (Fig. 3h), in comparison with the conventional Fenton process. Single ferrousions could reduce Cr(Ⅵ) (Fig. 3i), ascribed tothe redox reaction (Eq. 10) [31]. Apparently, the ferric ions were not beneficial to reduce Cr(Ⅵ). Similarly, Fe2+ +H2O2 system exhibits higher reduction activity than Fe3++H2O2 system, owing to the weak reducing capacity of Fe3+. And Fe3++H2O2 system shows a higher reducing activity than that of blank Fe3+, because of the reduction ability of H2O2 [32]. However, both Fe2+ +H2O2 and Fe3++H2O2 systems show an inhibition trend after 5 min, attributing to the loss of ferrous ions in suspension. Unsurprisingly, when MoO2 was added into the system, it was obviously found that the inhibition effect of Cr(Ⅵ) could be ended. The MoO2's surface is positively charged(pH ~4) (Fig.S8), leading to the adsorption of Cr2O72- on the surface of MoO2 due to the electrostatic effect. Hence, it can be proposed that the exposed metallic Mo4+ sites also can directly reduce Cr(Ⅵ) (Eq. 11). It is not strange that the Fe2+ +MoO2 system could reduce Cr(Ⅵ) more effectively than Fe2+ +H2O2+MoO2, owing to the reducing of Cr(Ⅵ) by the exposed metallic Mo4+. Although H2O2 has certain reducibility (Fig. S12 in Supporting information), its property is very unstable. Especially in the presence of Fe2+, H2O2 preferentially oxidizes Fe2+ into Fe3+ (Eq. 1), thus weakening the activity of Fe2+ in reducing Cr(Ⅵ). Therefore, under the presence of H2O2, Cr(Ⅵ) reduction is not necessarily promoted.

|
(10) |

|
(11) |
In summary, we have developed an inorganic catalytic system by MoO2 in Fenton process. With the aid of Mo4+ active metallic sites exposed on MoO2's surface, the efficiency of Fe(Ⅲ)/Fe(Ⅱ) cycle and the precipitation of iron sludge can be significantly enhanced and inhibited, respectively. Noticeably, it is found that 1O2 is responsible for the degradation of L-RhB by the cocatalytic effect of commercial MoO2 in Fenton reaction. Simultaneously, MoO2 cocatalytic AOPs can also achieve the reduction of Cr(Ⅵ). The MoO2 catalytic system not only achieves the goal of oxidizing organic pollutants, but also realizes the reduction of heavy metals. Thus, it can be expected that this work provides some guidance for 1O2 radicals dominated Fenton process and push forward the development of inorganic cocatalytic Fenton system for the mineralization of organic-inorganic contaminants to build a reliable platform for environmental remediation.
Declaration of competing interestThe authors declare that they have no conflicts of interest to this work.
AcknowledgmentsThis work was supported by the State Key Research Development Program of China (No. 2016YFA0204200), National Natural Science Foundation of China (Nos. 21822603, 21811540394, 5171101651, 21677048, 21773062, 21577036), Shanghai Pujiang Program (No. 17PJD011), and the Fundamental Research Funds for the Central Universities (No. 22A201514021). Project supported by Shanghai Municipal Science and Technology Major Project (No. 2018SHZDZX03) and the Program of Introducing Talents of Discipline to Universities (No. B16017). The authors thank to Research Center of Analysis and Test of East China University of Science and Technology for the help on the characterization.
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.09.052.
| [1] |
C. Dong, J. Ji, B. Shen, M. Xing, J. Zhang, Environ. Sci. Technol. 52 (2018) 11297-11308. DOI:10.1021/acs.est.8b02403 |
| [2] |
C. Dong, J. Lu, B. Qiu, et al., Appl. Catal. B-Environ. 222 (2018) 146-156. DOI:10.1016/j.apcatb.2017.10.011 |
| [3] |
E.C. Ryberg, C. Chu, J.H. Kim, Environ. Sci. Technol. 52 (2018) 13361-13369. DOI:10.1021/acs.est.8b03866 |
| [4] |
M. Xing, W. Xu, C. Dong, et al., Chem 4 (2018) 1359-1372. DOI:10.1016/j.chempr.2018.03.002 |
| [5] |
Y. Wang, F. Wicaksana, C.Y. Tang, A.G. Fane, Environ. Sci. Technol. 44 (2010) 7102-7109. DOI:10.1021/es101966m |
| [6] |
R.W. Baker, Kirk-Othmer Encyclopedia of Chemical Technology, John Wiley & Sons, Inc., Hoboken, 2005.
|
| [7] |
A. Jordan, N. Gathergood, Chem. Soc. Rev. 44 (2015) 8200-8237. DOI:10.1039/C5CS00444F |
| [8] |
D.W. Gao, Z.D. Wen, Sci. Total Environ. 541 (2016) 986-1001. DOI:10.1016/j.scitotenv.2015.09.148 |
| [9] |
X. Shi, Y. Wu, M. Wang, et al., Chem. Eng. Technol. 40 (2017) 847-853. DOI:10.1002/ceat.201600551 |
| [10] |
S.M. Louie, R.D. Tilton, G.V. Lowry, Environ. Sci-Nano 3 (2016) 283-310. DOI:10.1039/C5EN00104H |
| [11] |
B.F. Zheng, T. Ouyang, Z. Wang, et al., Chem. Commun. (Camb.) 54 (2018) 9583-9586. DOI:10.1039/C8CC04199G |
| [12] |
M.M. Hinojosa Guerra, I. Oller Alberola, Water Res. 156 (2019) 232-240. DOI:10.1016/j.watres.2019.02.055 |
| [13] |
Y.F. Fang, A.P. Deng, Y.P. Huang, Chin. Chem. Lett. 20 (2009) 1235-1240. DOI:10.1016/j.cclet.2009.05.004 |
| [14] |
X.T. Wang, T. Ouyang, L. Wang, et al., Angew. Chem. 58 (2019) 13291-13296. DOI:10.1002/anie.201907595 |
| [15] |
X. Yang, X. Cheng, A.A. Elzatahry, J. Chen, Chin. Chem. Lett. 30 (2019) 324-330. DOI:10.1016/j.cclet.2018.06.026 |
| [16] |
J. Qu, T. Che, L. Shi, Q. Lu, S. Qi, Chin. Chem. Lett. 30 (2019) 1198-1203. DOI:10.1016/j.cclet.2019.01.021 |
| [17] |
Y. Chen, S. Niu, W. Lv, C. Zhang, Q. Yang, Chin. Chem. Lett. 30 (2019) 521-524. DOI:10.1016/j.cclet.2018.04.019 |
| [18] |
Y. Pan, H. Su, Y. Zhu, H. Vafaei Molamahmood, M. Long, Water Res. 145 (2018) 731-740. DOI:10.1016/j.watres.2018.09.020 |
| [19] |
X. Dou, Q. Zhang, S.N.A. Shah, et al., Chem. Sci. 10 (2019) 497-500. DOI:10.1039/C8SC03511C |
| [20] |
J.M. Fontmorin, R.C. Burgos Castillo, Water Res. 99 (2016) 24-32. DOI:10.1016/j.watres.2016.04.053 |
| [21] |
Q. Yi, J. Ji, B. Shen, et al., Environ. Sci. Technol. 53 (2019) 9725-9733. DOI:10.1021/acs.est.9b01676 |
| [22] |
A.P. Grosvenor, B.A. Kobe, M.C. Biesinger, N.S. McIntyre, Surf. Interface Anal. 36 (2004) 1564-1574. DOI:10.1002/sia.1984 |
| [23] |
J. Hu, G. Chen, I.M.C. Lo, Water Res. 39 (2005) 4528-4536. DOI:10.1016/j.watres.2005.05.051 |
| [24] |
S.M. Ponder, J.G. Darab, T.E. Mallouk, Environ. Sci. Technol. 34 (2000) 2564-2569. DOI:10.1021/es9911420 |
| [25] |
T. Karthikeyan, S. Rajgopal, L.R. Miranda, J. Hazard. Mater. 124 (2005) 192-199. DOI:10.1016/j.jhazmat.2005.05.003 |
| [26] |
K. Selvi, S. Pattabhi, K. Kadirvelu, Bioresour. Technol. Rep. 80 (2001) 87-89. DOI:10.1016/S0960-8524(01)00068-2 |
| [27] |
P. Wang, I.M.C. Lo, Water Res. 43 (2009) 3727-3734. DOI:10.1016/j.watres.2009.05.041 |
| [28] |
Y. Wu, J. Zhang, Y. Tong, X. Xu, J. Hazard. Mater. 172 (2009) 1640-1645. DOI:10.1016/j.jhazmat.2009.08.045 |
| [29] |
J. Hu, I.M.C. Lo, G. Chen, Sep. Purif. Technol. 56 (2007) 249-256. DOI:10.1016/j.seppur.2007.02.009 |
| [30] |
J. Qiu, M. Li, L. Yang, J. Yao, Chem. Eng. J. 375 (2019) 121990. DOI:10.1016/j.cej.2019.121990 |
| [31] |
I.J. Buerge, S.J. Hug, Environ. Sci. Technol. 31 (1997) 1426-1432. DOI:10.1021/es960672i |
| [32] |
R. Ding, L. Qi, M. Jia, H. Wang, Electrochim. Acta 113 (2013) 290-301. DOI:10.1016/j.electacta.2013.09.053 |