 2019, Vol. 30
2019, Vol. 30 


b Department of Clinical Laboratory, The Affiliated Drum Tower Hospital of Nanjing University Medical School, Nanjing 210008, China;
c College of Life Science, Yangtze University, Jingzhou 434025, China;
d Bio Resources Conservation Institute, National Agriculture Research Centre, Islamabad 350000, Pakistan;
e Laboratory of Biointerface & Biomaterials, School of Biological Science and Medical Engineering, Southeast University, Nanjing 210096, China;
f Hunan Key Laboratory of Biomedical Nanomaterials and Devices, Hunan University of Technology, Zhuzhou 412007, China;
g National Center for International Bio-targeting Theranostics, Guangxi Key Laboratory of Bio-targeting Theranostics, Collaborative Innovation Center for Targeting Tumor Theranostics, Guangxi Medical University, Nanning 530021, China
Humans and dogs being friends shared a long history. Worldwide people keep dogs for various purposes. Pet dogs are considered as members of the family [1, 2]. CPV-2, a member of Parvovirus genus, specifically infect non vaccinated puppies (6–12 weeks), and manifests itself in the form of gastroenteritis [3, 4]. CPV-2 genome is a linear single-stranded DNA molecule of about 52 kb in size, encapsulated in a non-enveloped capsid of about 25 nm diameter [5, 6]. Since 1978, CPV-2 has been a cause of haemorrhagic gastroenteritis in dogs, with high rate of mortality and morbidity worldwide [7]. Therefore, early diagnosis, management and monitoring of CPV-2 infection in dogs are pertinent.
Clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR-associated (CRISPR-Cas) systems are adapted form a bacterial adaptive immune system [8, 9]. CRISPR/Cas system is used to degrade foreign nucleic acids entering the host cell [10, 11]. Cas13a (old name C2C2) has been characterized as an RNAguided RNA manipulating enzyme that harnesses single-stranded CRISPR RNA (crRNA) with a spacer region at 3 prime-terminal for target binding and subsequent editing [12, 13]. Cas13a-crRNA complex when binds the target RNA it performs conformational changes that results in nonspecific RNA degradation including target RNA and other surrounding RNA. During the cleavage of target RNA, Cas13a cleaves ~104 nonspecific RNA as well [12-14]. This collateral cleavage activity of Cas13a can be reined in for the sensitive detection of RNAs. SHERLOCK has been successfully reported for the on-site detection of deadly human viruses like Zika, Dengue and Ebola [10, 15, 16].
Over the years, CPV-2 sensitive and specific diagnosis has been of interest for scientists [1]. Many methods have been reported previously [17-23], but they lack specificity, sensitivity, time consuming and needs sophisticated instruments. These issues are bottleneck in their widespread application for on-site detection. Therefore, to accomplish on-site detection of CPV-2 DNA, we employed SHERLOCK [15]. CPV-2 genome was used as target spacer for corresponding crRNA to establish a new CRISPR-Cas13a based detection system for CPV-2. To boost the sensitivity of our method RPA was also employed. The process does not require expensive equipment as well it is less laborious and time-efficient, the prerequisites for an excellent detection system.
The pC013-Twinstrep-SUMO-huLwCas13a (Addgene plasmid #90097) was gifted by Broad Institute Zhang Feng Laboratory. CPV-2 and other types of viruses were provided by Professor Xiong Tao's laboratory, College of Life Science, Yangtze University. Primers and quenched fluorescent probe were synthesized by Genscript (Jiangsu, China) and Sangon Biotech (Shanghai, China). Oligonucleotides engaged in this study are shown in Table 1.
|
|
Table 1 Oligonucleotides used in this study. |

Mammalian codon optimized LwCas13a gene (encoding 11152 residues) was synthesized (Genescript) and subsequently cloned into bacterial expression system, pC013 Twinstrep-SUMOhuLwCas13a (Addgene plasmid #90097) to obtain the recombinant protein Cas13a. Using these plasmids about 50 ng, BL21 E. coli competent cells were transformed (Cwbiotech). After transformation 100 μL cells were plated on ampicillin and chloramphenicol LB-agar plate for 12–15 h at 37 ℃. Afterwards colonies were counted. Single colony was cultured in 10 mL starter LB medium with ampicillin to a final concentration of 100 μmol/L overnight. That was further used to inoculate 500 mL of LB medium with 100 μmol/L of ampicillin for growth at 37 ℃ and 220 RPM until 0.6 optical density (OD). Once the concentration met the requirements, isopropyl β-D-1-thiogalactopyranoside (IPTG) with a final concentration of 500 μmol/L was added to induce protein expressionwhile adjusting incubator temperature to 16 ℃. After 16 h of cultivation, cells were then centrifuged at 4500×g for 10 min at 4 ℃. Cell pellet was harvested and re-suspended in lysis buffer (20 mmol/L Tris-HCl, 500 mmol/L NaCl, 1 mmol/L DTT, pH 7.4), supplemented with protease inhibitors (MCE) and lysozyme, followed by sonication (JX-650, Shanghai, China) by adjusting conditions as: 200 W of power, ultrasonic 2.5 s and 10 s of pause for 25 min. The soluble substances were separated from insoluble impurities by centrifugation for 90 min at 4 ℃ at 10, 000×g, and the supernatant was filtered through a 220 nm filter (Sterile Millex). SUMO protease was then loaded onto the column and incubated overnight at 4 ℃. The recombinant proteins were further purified by 5 mL HisTrap HP nickel ion affinity column by removing the 6 His-SUMO tag using AKTA purifier (AKTA PURE, GE Healthcare Life Sciences) and then the column was washed by wash buffer (20 mmol/L Tris-HCl, 500 mmol/L NaCl, 30 mmol/L imidazole, pH 7.4). The purity of the recombinant proteins was evaluated by SDS-PAGE. Fractions containing pure LwCas13a were pooled and buffer exchanged via a Millipore centrifugal ultrafiltration tube (Amicon Ultra) to storage buffer (600 mmol/L NaCl, 50 mmol/L Tris-HCl, pH 7.5, 5% glycerol, 2 mmol/L DTT) and kept at -80 ℃.
Sample of DNA collected from the dog's intestine using DNeasy kit was PCR amplified with Prime STAR Max DNA Polymerase (TaKaRa). Purified DNA was then transcribed by T7 RNA polymerase into an RNA copy using HiScribe T7 kit (New England Biolabs) according to the manufacturer's protocol, and was purified with RNA Rapid Concentration Purification Kit (Sangon Biotech). CrRNA was obtained using artificially synthesized oligonucleotide primers with self-complementary regions having an appended T7 promoter sequence. Two complementary pairs of crRNA DNA primers (final concentrations 10 μmol/L) were annealed (Solarbio) and incubated with T7 polymerase overnight at 37 ℃ using the HiScribe T7 Quick High Yield RNA Synthesis Kit (New England Biolabs). CrRNAs were then purified using RNA Rapid Concentration Purification Kit (Sangon Biotech).
Specificity of LwCas13a was determined according to a published protocol with slight modifications [15]. Briefly we made three samples with 1 μL murine RNase inhibitor (YEASEN), 45 nmol/L purified LwCas13a, 22.5 nmol/L crRNA and 50 nmol/L target DNA (Sangon Biotech, New England Biolabs) and measured its fluorescence through SpectraMax M5. Briefly, 100 ng of background total human RNA (purified from monocytes, more accurate measurement of trans-cleavage activity in the presence of total RNA), and 1.25 μmol/L quenched fluorescent RNA probe (Genscript) were mixed in NAB buffer (40 mmol/L Tris-HCl, 60 mmol/L NaCl, 6 mmol/L MgCl2, pH 7.3) to 20 μL final volume. For negative control groups once target RNA was replaced with non-target and Cas13a protein with non-Cas13a protein respectively.
The reporter RNA molecule is an oligonucleotide beacon with a fluorescent dye sitting on its 50-end and a quencher on its 30-end. When the molecule is cleaved, the dye is separated from the quencher emitting light in the green spectrum at 520 nm wavelength. The quenched fluorescent RNA probe was incubated with RNase to confirm that the probe could act as a signal molecule. Reactions were incubated in a 384-well microplate format, which was then detected in a fluorescence plate reader (SpectraMax M5) for up to 30 min at 37 ℃ before fluorescence measurements (λex: 492 nm; λem: 518 nm).
Improved detection assays were performed with RPA (TwistDx) and T7 transcription in vitro transcription (New England Biolabs) as follows: 0.48 μmol/L forward primer, 0.48 μmol/L reverse primer, 10 μL 2X Reaction Buffer, 1.8 mmol/L dNTP, 1 μL murine RNase inhibitor, 45 nmol/L purified LwCas13a, 22.5 nmol/L crRNA, 100 ng of background total human RNA, 2 mmol/L rNTP, 0.5 μL T7 Mix, 5 mmol/L MgCl, 2 μL 10X Basic E-mix, 1 μL 20X Core Reaction Mix, 0.4 μL 50X RT, 14 mmol/L MgOAc and various concentrations of target DNA to 20 μL final volume. Reactions were then incubated in a 384-well microplate format, which was detected in a fluorescence plate reader for up to 90 min at 37 ℃ with fluorescence measurements taken every 5 min (λex: 492 nm; λem: 518 nm).
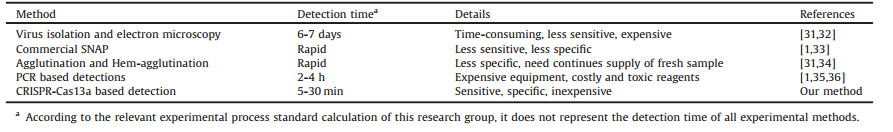
Previously LwCas13a protein had been purified in a complex multi-step process [15, 24]. However, same approach was not used in our study because of expensive instruments and columns. Hereby we developed new LwCas13a purification method based on SUMO enzyme digestion. SDS-PAGE was used to check prokaryotic LwCas13a expression (Fig. 1). LwCas13a molecular weight along with SUMO site was 155.2 kDa while the molecular weight of digested LwCas13a by SUMO enzyme was 138.5 kDa. The Coomassie blue-stained gel results show that E. coli treated with IPTG shows higher protein expression than E. coli without IPTG treatment (Figs. 1A and B). As IPTG is expensive as well cytotoxic reagent so we wanted to optimize IPTG concentration for LwCas13a induction. We found that lower final concentration of IPTG (0.1–0.6 mmol/L) has significantly induced the expression of LwCas13a comparatively (Fig. 1C).

|
Download:
|
| Fig. 1. SDS-PAGE of recombinant LwCas13a. (A) M: protein marker, 1: uninduced E. coli, 2: E. coli induced by IPTG. (B) M: protein marker, 1: E. coli not induced by IPTG, 2: E. coli induced by IPTG, 3: supernatants after disruption, 4: insoluble impurities. (C) IPTG follow-through for induction of Cas13a. | |
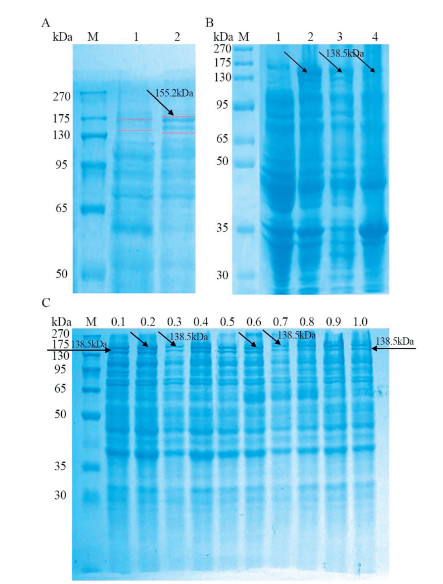
After ultra-sonication of E. coli cells, we found that LwCas13a was mainly present in supernatants of the solution. The protein purification as shown in Fig. 2, was done via nickel ion affinity chromatography (Fig. 2A) and confirmed by running SDS PAGE for the purified protein (Fig. 2B). Further different volumes of purified fluid were pooled and buffer exchanged via a Millipore centrifugal ultrafiltration tube to the storage buffer.

|
Download:
|
| Fig. 2. Cas13a purification results. (A) AKTA PURE results of nickel ion affinity chromatography for Cas13a elution (Cas13a protein elution peak at 4.88 mL). (B) SDS-PAGE analysis of purified Cas13a (Lane M: 270 kDa protein marker, Lane 1: Purified Cas13a). | |
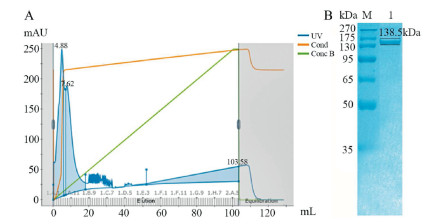
The obtained LwCas13a protein was tested for non-specific RNase activity while using RNase A as positive reference. The background-subtracted fluorescence was significantly lower in the absence of LwCas13a or crRNA or target RNA compared to the intact reaction system (Fig. 3A) indicating that the Cas13a had the ability to non-specifically cleave RNA in the presence of target RNA and its corresponding crRNA. Various concentrations of target nucleic acid were then added into the reaction system. The background-subtracted fluorescence of each reaction system was quantitatively related to the target RNA concentration (between 100 pmol/L to 100 nmol/L) in the system at 30 min.

|
Download:
|
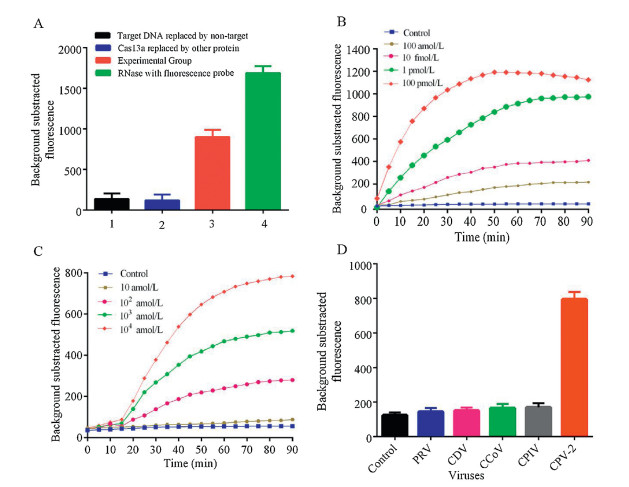
| Fig. 3. (A) Activity detection of LwCas13a. Bar 1: Target RNA replaced by non-target; Bar 2: Cas13a replaced by another protein; Bar 3: Experimental group; Bar 4: RNase A with quenched florescence probe. (B) Concentration gradient detection of CPV-2 DNA. (C) CPV-2 minimum detection and time limit through CRISPR-Cas13a detection system. (D) Detection of various dog viruses with optimized Cas13a detection system (n = 3 technical replicates, Bars represents mean ± S.E.M). | |
As CPV-2 is an ssDNA virus so we cannot detect CPV-2 directly through CRISPR-Cas13a detection system therefore, we have converted DNA to RNA. We employed RPA and T7 transcription for the amplification of our nucleic acid target. RPA and T7 transcription system also improved the sensitivity of the detection system. Our detection system consisted of 0.48 μL RPA forward primer, 0.48 μL RPA reverse primer, 10 μL 2X Reaction Buffer, 1.8 mmol/L dNTP, 1 μL RNase inhibitor, 2 μL 10X Basic E-mix, 1 μL 20X Core Reaction mix, 0.4 μL 50X RT, 14 mmol/L MgOAc, 5 μmol/L RNA probe, 2.5 μL. Different concentration of the target DNA and finally buffer was used to make the final volume up to 20 μL. These samples were then placed in a 384 well plate and were taken to SpectraMax5 for reading (excitation wavelength was 492 nm and emission wavelength was 518 nm). The system was allowed to run for 1 h and 30 min and readings were obtained after every 5 min (Fig. 3B). We diluted some of the CPV DNA into 10 fmol/L sample and after we made a 10-fold serial dilution of this sample, followed by the addition of Cas13a protein. The readings obtained after detecting the sample through spectraMax5 are shown in Fig. 3C. The optimized identification system was able to detect 100 amol/L of CPV-2 DNA within 30 min.
Specificity is an important indicator for evaluating the detection system. As symptoms of CPV-2 infection are similar to those of most other dog's viruses, that why for verification of Cas13a based nano-detection system specificity we used several other dog's viruses, including pseudorabies virus (PRV, PRV-R1 strain), canine distemper virus (CDV, CDV-NJ2 strain), canine coronavirus (CCoV, CCoV-C5 strain), canine parainfluenza virus (CPIV, CPIV-J2 strain). The results showed that the average fluorescence value for CPV-2 was significantly higher compared to PRV, CDV, CCoV, CPIV (Fig. 3D), which indicates that crRNA along with RPA primers have good specificity for CPV-2 and provides proof that Cas13a based nanosystem can be used specifically for CPV-2 detection.
Several published papers reported methods for the detection of CPV-2 [25-30]. CRISPR-Cas13a nanosystem when compared with previously reported detection platforms (Table 2 [1, 31-36]) it can detect CPV-2 DNA at 100 amol/L level in 30 min. The salient feature of the proposed system is it being time-efficient, cost-effective, sensitive, and avoiding the use of sophisticated lab instruments that need separate room and sterile conditions. This nanosystem can be used for both research and diagnostic purpose. In short, the CRISPR-Cas13a nano-detection system is suitable for on-site detection of CPV-2 which will contribute to the future control of CPV-2 outbreaks.
|
|
Table 2 Comparison of our method with previous methods used for CPV detection. |
{kind=link}
{kind=link}
{kind=link}
Declaration of competing interest
The authors declare that they have no conflict of interest.
AcknowledgmentsThis work was supported by the National Key Research and Development Program of China (No. 2017YFA0205301), National Natural Science Foundation of China (Nos. 81902153, 61527806 and 81430055), Key Research and Development Project of Jiangsu Province (No. BE2019761), Programs for Changjiang Scholars and Innovative Research Team in University (No. IRT_15R13) and open Funding of State Key Laboratory of Oral Diseases (No. SKLOD2019OF03).
| [1] |
H. Cun, P. Miguel, R. Armenio, et al., Hum. Relat. 72 (2019) 778-800. DOI:10.1177/0018726718780210 |
| [2] |
A. Treves, C. Bonacic, Trends Ecol. Evolut. 31 (2016) 489-491. DOI:10.1016/j.tree.2016.04.006 |
| [3] |
Q.Y. Zhuang, Y. Qiu, Z.H. Pan, et al., Transbound. Emerg. Dis. 66 (2019) 897-907. DOI:10.1111/tbed.13100 |
| [4] |
J.D. Figueire, C. Miranda, R. Souto, et al., Arch. Microbiol. 199 (2017) 543-549. DOI:10.1007/s00203-016-1320-7 |
| [5] |
H. Wu, X. Li, L. Wang, et al., Virus. Dis. 29 (2018) 113-117. DOI:10.1007/s13337-018-0427-7 |
| [6] |
C. Li, J. Tang, Z. Chen, et al., Infect. Genet. Evol. 73 (2019) 242-247. DOI:10.1016/j.meegid.2019.05.004 |
| [7] |
D. de la Torre, P.B. Mafla, L. Erazo, et al., Vet. World 11 (2018) 480-487. DOI:10.14202/vetworld.2018.480-487 |
| [8] |
C. Janic, M. Enbo, H. Lucas, et al., Science 360 (2018) 436-439. DOI:10.1126/science.aar6245 |
| [9] |
L. Cong, F.A. Ran, D. Cox, et al., Science 339 (2013) 819-823. DOI:10.1126/science.1231143 |
| [10] |
P. Qin, M. Park, K.J. Alfson, et al., ACS Sens. 4 (2019) 1048-1054. DOI:10.1021/acssensors.9b00239 |
| [11] |
R. Jansen, J.D.A. Embden, W. Gaastra, et al., Mol. Microbiol. 43 (2002) 1565-1575. DOI:10.1046/j.1365-2958.2002.02839.x |
| [12] |
L. Liu, X. Li, J. Ma, et al., Cell 170 (2017) 714-726. DOI:10.1016/j.cell.2017.06.050 |
| [13] |
O.O. Abudayyeh, J.S. Gootenberg, S. Konermann, et al., Science 353 (2016) aaf5573. DOI:10.1126/science.aaf5573 |
| [14] |
A.E. Seletsky, A. O'Connell, M.R. Knight, et al., Nature 538 (2016) 270-273. DOI:10.1038/nature19802 |
| [15] |
J.S. Gootenberg, O.O. Abudayyeh, J.W. Lee, et al., Science 356 (2017) 438-442. DOI:10.1126/science.aam9321 |
| [16] |
A.A. Hatoum, Clin. Chem. 64 (2018) 1681-1683. DOI:10.1373/clinchem.2018.295485 |
| [17] |
K. Uwatoko, M. Sunairi, M. Nakajima, et al., Vet. Microbiol. 43 (1995) 315-323. DOI:10.1016/0378-1135(94)00102-3 |
| [18] |
T.X. Castro, C.M.A. Uchoa, M.C. Albuquerque, Int.J.Appl.Res.Vet.Med. 5 (2007) 72-76. |
| [19] |
M. Mochizuki, M.V.S. Gabriel, H. Nakatani, et al., Res. Vet. Sci. 55 (1993) 60-63. DOI:10.1016/0034-5288(93)90035-E |
| [20] |
Y. Suna, Y. Chenga, P. Lina, et al., Mol. Cell. Probes 38 (2018) 7-12. DOI:10.1016/j.mcp.2018.02.004 |
| [21] |
C. Desario, N. Decaro, M. Campolo, et al., J. Virol. Methods 126 (2005) 179-185. DOI:10.1016/j.jviromet.2005.02.006 |
| [22] |
Y. Geng, J. Wang, L. Liu, et al., BMC Vet. Res. 13 (2017) 311. DOI:10.1186/s12917-017-1232-z |
| [23] |
S.R. Kersting, V. Rausch, F.F. Bier, M.N. Rosenegk, Microchim. Acta 181 (2014) 1715-1723. DOI:10.1007/s00604-014-1198-5 |
| [24] |
J.S. Gootenberg, O.O. Abudayyeh, M.J. Kellner, et al., Science 360 (2018) 439-444. DOI:10.1126/science.aaq0179 |
| [25] |
C. Desario, N. Decaro, M. Campolo, et al., J. Virol. Methods 126 (2005) 179-185. DOI:10.1016/j.jviromet.2005.02.006 |
| [26] |
J. Wang, C. Liu, R. Li, Arch. Virol. 161 (2016) 1015-1018. DOI:10.1007/s00705-015-2738-y |
| [27] |
H.K. Mukhopadhyay, S. Amsaveni, S.L. Matta, et al., Lett. Appl. Microbiol. 55 (2012) 202-209. DOI:10.1111/j.1472-765X.2012.03284.x |
| [28] |
V. Gupta, S. Chakravarti, V. Chander, et al., Arch. Virol. 162 (2017) 1995-2001. DOI:10.1007/s00705-017-3321-5 |
| [29] |
R.K. Daher, G. Stewart, M. Boissinot, M.G. Bergeron, Clin. Chem. 62 (2016) 947. DOI:10.1373/clinchem.2015.245829 |
| [30] |
J. Wang, J. Wang, R. Li, et al., BMC Vet. Res. 13 (2017) 241. DOI:10.1186/s12917-017-1180-7 |
| [31] |
S.S. Jevdenic, D. Trailovic, B. Vidic, M. Jovanovic, Acta Veterinaria-Beograd 56 (2006) 515-527. DOI:10.2298/AVB0606515S |
| [32] |
A.F. Ortega, J.S. Martinez-Castaneda, L.G. Bautista-Gomez, et al., Braz. J. Microbiol. 48 (2017) 769-773. DOI:10.1016/j.bjm.2017.03.008 |
| [33] |
L.B. Liu, J.C. Wang, Y.Y. Geng, et al., Mol. Cell. Probes 39 (2018) 41-46. DOI:10.1016/j.mcp.2018.04.004 |
| [34] |
J. Thomas, M. Singh, T.K. Goswami, et al., Biologicals 49 (2017) 51-56. DOI:10.1016/j.biologicals.2017.06.009 |
| [35] |
M. Mochizuki, S. Gabriel, M.C. Nakatani, et al., Res. Vet. Sci. 55 (1993) 60-63. DOI:10.1016/0034-5288(93)90035-E |
| [36] |
Q. Fan, Q. Xie, Z.X. Xie, et al., J. Virol. Methods 186 (2012) 43-48. DOI:10.1016/j.jviromet.2012.08.007 |