 2019, Vol. 30
2019, Vol. 30 


b University of Chinese Academy of Sciences, Beijing 100049, China;
c The Key Laboratory of Water and Sediment Science, Ministry of Education, College of Environmental Sciences and Engineering, Peking University, Beijing 100871, China;
d The Beijing Innovation Center for Engineering Science and Advanced Technology(BIC-ESAT), Peking University, Beijing 100871, China;
e Beijing Engineering Research Center for Advanced Wastewater Treatment, Peking University, Beijing 100871, China
Recent years, as a class of emerging contaminants, pharmaceuticals and personal care products (PPCPs), such as antidepressants, antibiotics and anti-inflammatories, have drawn extensive concerns [1-3]. PPCPs are widely detected in water and wastewater matrix and show high potential toxicity risks to aquatic life/human beings [1-3]. Nonsteroidal anti-inflammatory drugs (NAIDS) are one of the most widely available and consumed pharmaceuticals with 70 million annual prescriptions in the world [4-6]. Ibuprofen (2-[3-(2-methylpropyl)phenyl]propanoic acid, IBP), which belongs to a class of NAIDS, has been reported that its mean concentration in ground, surface and drinking water is up to 0.024, 0.24 μg/L and 5–25 ng/L, respectively [7-9]. Although the detected concentration of IBP is low, it still holds unexpected toxicity to eco-system and human health [10]. Moreover, the conventional treatment technologies for drinking water, such as coagulation, sedimentation, filtration and disinfection, are not efficient and suitable to remove IBP at low concentration levels [11]. Thus, it is urgent to develop efficient strategies for IBP removal.
Heterogenous catalysis using functional materials has been widely applied for organics removal [12-16], especially the radicalinvolved processes. Although the hydroxyl radical (·OH, E0 = +1.8– 2.7 V) exhibits strong power to oxidize organics, the sulfate radical (SO4·-, E0 = +2.5–3.1 V) has some unique and innovative potentials, i.e., higher selectivity, longer radical half-life (30–40 μs for SO4·- and 1 ms for ·OH) and wider pH application range [17, 18]. Previous works developed many technologies to activate peroxymonosulphate (PMS, HSO5-) and persulphate (PS, S2O82-) for generating sulfate radicals, such as degradation of p-nitrophenol by mFe/Cuair-PS activation system [13], dye pollutants degradation by CuFe2O4@GO-PMS activation system [19], organic pollutants removal by sponge of cobalt heterostructures-PMS [20]. The Co-modified materials have been believed the most efficient heterogeneous or homogeneous catalysts to activate PMS or PS [21].
The overall goal of this study was to develop and test the effectiveness of a hollow structure Co(OH)2 material to activate PMS for ibuprofen degradation. The specific objectives were to: 1) develop an optimized solvothermal-hydrothermal method to prepare the hollow Co(OH)2, 2) investigate the removal efficiencies of IBP by various PMS activation systems, 3) reveal the IBP degradation mechanism and radical attacking function based on degradation intermediate/products identification and computational chemistry analysis, 4) assess the effects of water chemistry conditions including pH and PMS concentration, and 5) elucidate the underlying PMS efficient activation mechanism through material characterizations.
All chemicals used in this work were of analytical grade or higher. Details related to the chemicals are provided in Text S1 (Supporting information). Hollow structure cobalt hydroxide (hCo(OH)2) were prepared through a modified solvothermalhydrothermal method [22]. Specifically, 10 mmol/L (2.9105 g) Co(NO3)2·6H2O was dissolved in 60 mL isopropanol and then 16 mL glycerin was added. The mixture was magnetically stirred for 30 min to completely dissolved and then transferred into a Teflon reactor with stainless steel coating, heated at 180 ℃ for 6 h to complete solvothermal reaction. After cooling to the room temperature, the pink precipitates were washed with ethanol for 3 times to remove the residuals anions and oven-dried at 60 ℃ for 12 h to obtain alkoxy cobalt microspheres (s-CoA). Another hydrothermal reaction was initiated then. 0.5 g s-CoA was dissolved in 80 mL deionized (DI) water, and the solution was transferred to the Teflon reactor and heated at 160 ℃ for 3 h. After cooling, the light green precipitates were washed with DI water for 3 times and dried at 60 ℃ for 12 h. Finally, the hollow structure cobalt hydroxide were ground and collected. For comparison, neat Co(OH)2 was also prepared via a conventional precipitation method (Test S2 in Supporting information) and labeled as p-Co (OH)2.
Batch kinetic experiments were carried out to test the effectiveness of h-Co(OH)2 for IBP degradation after PMS activation. The initial IBP concentration was fixed at 10 μmol/L. In a typical PMS activation system, 0.2 mmol/L PMS and solution pH were adjusted to 7.0 using diluted HClO4 (0.1 mmol/L) or NaOH (0.1 mmol/L). The mixture was then magnetically stirred at 300 rpm, and the heterogeneous catalytic reaction was initiated by adding 0.2 g/L cobalt material. At pre-determined time, 1 mL sample was collected and immediately filtrated via a 0.22 μm polytetrafluoroethylene (PTFE) membrane, which was pre-filled with 0.1 mL of 0.2 mmol/L Na2SO3 solution to quench the residual PMS-induced radicals. Control tests were conducted without any catalysts (estimate the degradation by PMS) or without PMS (quantify the adsorption by materials) but under otherwise identical conditions. IBP concentration in the filtrate was measured by a high-performance liquid chromatography (HPLC) system (Agilent 1260 Infinity, USA). The intermediates and products after IBP degradation were analyzed on a high-performance liquid chromatography-mass spectroscopy system (HPLC-MS, HP 1100 LC-MSn Trap SL System, Agilent). The details are shown in the Text S3 (Supporting information).
For pH effect, the solution pH was adjusted from 3 to 11. To quantify the contributions of different reactive oxygen species (ROS) to IBP degradation, scavengers including tert-butanol (tBA) or ethanol (2 mmol/L) was added before reaction to quench hydroxyl radical (·OH) and all radicals, respectively.
To evaluate the regioselectivity of generated radicals in the heterogeneous catalytic process for IBP molecules attacking, Fukui function (f-) based on the density functional theory (DFT) calculation was applied. Details on DFT calculation are presented in the Text S4 (Supporting information).
The materials were characterized by transmission electron microscopy (TEM), X-ray photoelectron spectroscopy (XPS), and powder X-ray diffraction (XRD). Meanwhile, the surface area and pore size distribution of the materials were obtained by the Brunauer-Emmett-Teller (BET) and the Barrett-Joyner-Halenda (BJH) method (Text S5 in Supporting information).
Fig. 1 depicts the TEM images of h-Co(OH)2. Unlike the p-Co (OH)2 prepared by directly conventional precipitation method, which generally exhibits broken and uneven flat platelets in the 100–500 nm range in size [23], h-Co(OH)2 was observed as hollow structure (Fig. 1a) and totally transformed into aggregated nanosheet-shell-like clusters after solvothermal-hydrothermal reaction (Fig. 1b). In addition, h-Co(OH)2 were microscale hollow spheres (Fig. 1b), in which consist of the center holes of ~1 mm and thin nanosheets shell of ~10 nm (Figs. 1c and d). TEM analysis indicated that the nanosheets-shell had crystalline characteristic (Fig. 1d), which is further confirmed by the XRD analysis. In addition, the h-Co(OH)2 with hollow structure exhibits larger specific surface area (132.36 m2/g) and total pore volume (0.57 cm3/g) than that of the p-Co(OH)2 (23.9 m2/g and 0.088 cm3/g) [24] (Table S1, Figs. S1 and S2 in Supporting information), suggesting efficient interaction with PMS for its activation by h-Co(OH)2.

|
Download:
|
| Fig. 1. TEM images of h-Co(OH)2. Scale bars: 2 μm (a), 1 μm (b), 50 nm (c), 20 nm (d). | |
{kind=link}
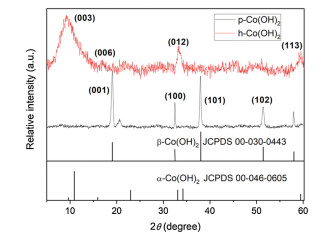
Fig. 2 presents XRD patterns of p-Co(OH)2 and h-Co(OH)2. For conventional p-Co(OH)2, four distinct peaks at 19.1°, 32.5°, 38° and 51.4° appeared and assigned to the crystalline plane (001), (100), (101) and (102) of β-Co(OH)2, respectively (JCPDS 00-030-0443) [23, 25, 26]. However, all the significant characteristic peaks belonged to α-Co(OH)2 in the h-Co(OH)2 spectra, as 9.5°, 17.8°, 33.7° and 59.5° peaks were assigned to the (003), (006), (012) and (113) crystalline plane of α-Co(OH)2 (JCPDS 00-046-0605), respectively [23, 27]. It is indicated that different crystalline phase formed for h-Co(OH)2 compared with conventional p-Co(OH)2 due to the solvothermal-hydrothermal method. In addition, the different Co(OH)2 phases can contribute to the distinct crystalline plane (reactive sites) to active PMS and result in the different removal efficiency of IBP.

|
Download:
|
| Fig. 2. XRD patterns of p-Co(OH)2 and h-Co(OH)2. | |
{kind=link}
Fig. S3 (Supporting information) shows XPS spectra of p-Co (OH)2 and h-Co(OH)2. For these two materials, the significant peaks of and O 1s in the survey spectra (Fig. S3a) indicated the similar element composition. In the high resolution of Co 2p spectra (Fig. S3b), the two group peaks at 781.1 and 796.8 eV for both materials are attributed to Co2+ in Co(OH)2 [23, 26]. The existence of satellite vibration peaks near 786.2 eV and 802.6 eV for Co 2p3/2 and Co 2p1/2 confirms the formation of Co3+ due to surface oxidation [28, 29].
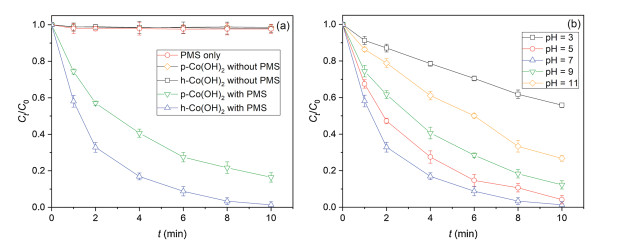
Fig. 3 shows the removal of IBP by PMS activated by h-Co(OH)2 or p-Co(OH)2. Control tests indicated that almost no removal (< 0.3%) of IBP by PMS direct degradation. Moreover, the adsorption of IBP by both h-Co(OH)2 and p-Co(OH)2 was negligible (< 0.2%), it is because the inorganic structure of Co(OH)2 is not sufficient to absorb organic compounds, which is consistent with previous reports [30, 31]. However, rapid and high degradation (83.6% and 98.6%) of IBP was observed within 10 min for both p-Co(OH)2 and h-Co(OH)2 with PMS, respectively, indicating the observed IBP degradation can be solely contributed to the efficient PMS activation by h-Co(OH)2 or p-Co(OH)2. The pseudo-first order kinetic model is used to interpret the kinetic data (Eq. (1)) [32-34]:
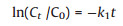
|
(1) |

|
Download:
|
| Fig. 3. IBP removal kinetics in various systems (a) and effects of pH on IBP degradation by h-Co(OH)2 activated PMS (b). (Initial IBP =10 μmol/L, material dosage = 0.2g/L, PMS concentration = 0.2mmol/L, initial pH 7.0). | |
{kind=link}
where C0 and Ct are the IBP concentrations (μmol/L) at time 0 and t (min) in aqueous phase, respectively; and k1 is the first-order rate constant (min-1).
Fig. S4 (Supporting information) shows the linear model fitting to IBP degradation in the heterogenous catalysis systems with PMS and Co(OH)2, and Table S2 (Supporting information) summarizes the best-fitted parameters. The pseudo-first order kinetic model can well describe the kinetic data (R2 >0.966). In addition, the rate constant (k1) increased from 0.195 min-1 for p-Co(OH)2 to 0.428 min-1 for h-Co(OH)2, by 2.2 times, indicating higher reactivity of h-Co(OH)2 for PMS activation. It can be attributed to the different morphology and crystalline phases as aforementioned (Figs. 1 and 2). Therefore, in the following experiments and characterizations, h-Co(OH)2 was focused.
The mechanism on activation of PMS by Co(OH)2 for radicals production and organics degradation can be summarized as Eqs. (2)–(10) [30, 31, 35, 36]:
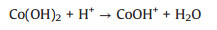
|
(2) |

|
(3) |
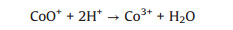
|
(4) |

|
(5) |

|
(6) |

|
(7) |
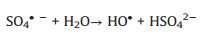
|
(8) |
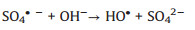
|
(9) |

|
(10) |
The formation of CoOH+, which has been reported as the key cobalt species to activate PMS [31] as shown in Eq. (2), which is the rate-limiting step for radical production [37]. After decomposition of PMS to generate SO4·-, the CoOH+ will transfer into CoO+ (Eq. (3)) and then further react with H+ to produce Co3+ (Eq. (4)). PMS can also consume Co3+ to regenerate Co2+ (Eq. (5)), which is the critical process to maintain the chain reactions at a relative low cobalt concentration [38]. And then the regenerated Co2+ can either quench the SO4·- (Eq. (6)) or reproduce the most efficient activation specie CoOH+ for reactivating PMS (Eq. (7)). It is worth noting that the generated SO4·- also can be captured by H2O or OH- for formation of ·OH (Eqs. (8) and (9)), which is also a strong radical for organics degradation. The generated sulfate radicals will further attack the active sites of IBP molecules for its degradation or mineralization (Eq. (10)).
Fig. 3b presents the effect of pH on IBP degradation in the PMS catalytic system. Increasing pH from 3 to 7 increased the k1 value from 0.059 min-1 to 0.428 min-1, and the removal efficiency increased from to 44.2% to 98.6% after 10 min reaction. At low pH, PMS with a pKa of 9.4 exists in the form of H2SO5 [39], and the formation of key cobalt species CoOH+ is inhibited due to acid condition [40, 41], so low efficiency is obtained. While at higher pH, SO4·- will react with OH- to form ·OH (Eq. (9)), which has low oxidation capacity compared to SO4·- at alkaline conditions [42]. Therefore, pH 7 is the optimum pH for PMS activation and then IBP degradation in this system.
Leaching of Co ion into solution during the reaction was also evaluated (Table S3 in Supporting information). It is found that only 2.36% of Co was dissolved into solution at pH 7 after 10 min, because the Co species transformation cycle shown in Eqs. (2)–(7). In addition, it is worth noting that the PMS activation reaction by hCo(OH)2 is a self-sacrificing process, so reusability of the catalyst in this advanced oxidation process is generally limited [29, 31, 43]. In the future, developing new catalysts or new strategies to ensure the metal cycle is the key issues for this area.
To further identify the contributions of main radicals involved in the IBP degradation, classical scavenger quenching tests were conducted. Ethanol [33, 44] and tBA [45-47] was applied as the scavenger of ·OH and all radicals including ·OH and SO4·-, respectively (Fig. 4). Table S2 lists the rate constant (k1) by for IBP degradation in the presence of various scavengers. After tBA and ethanol added, IBP removal was inhibited, as the k1 value decreased from 0.428 min-1 (without scavenger) to 0.261 min-1 and 0.020 min-1, respectively (Table S2). In addition, for the catalytic system using h-Co(OH)2 at 10 min, ·OH contributed 6.3% while SO4·- contributed 75.7% to the IBP removal, respectively. Luo et al. [29] and Yun et al. [48] have mentioned the singlet oxygen (1O2) may dominate the non-radical oxidation process in the PMS activation. Thus, another quenching test for 1O2 was conducted using NaN3. However, the contribution of 1O2 was negligible (< 2%), which was not the primary reactive species in this activation system. Therefore, the dominant mechanism for the IBP removal by PMS after h-Co(OH)2 activation in this study is radical-driven processes due to SO4·-, and ·OH demonstrated a less contribution. The details on radical attacking mechanism will be discussed in the following paragraph based on the DFT computational analysis.

|
Download:
|
| Fig. 4. Effects of reactive species for IBP removal by h-Co(OH)2 activated PMS. (Initial IBP =10 μmol/L; material dosage = 0.2 g/L; PMS concentration = 0.2 mmol/L; initial pH = 7.0; ethanol, tBA and NaN3 concentration = 2 mmol/L). | |
{kind=link}
SO4·- and ·OH are the primary attack reactive oxygen species in this system, and both of them are classified as a kind of electrophilic radicals, which are more likely to attack the sites that can readily lose electron [49]. Therefore, Fukui index (f–) indicating electrophilic attack is considered and Figs. 5a and b displays the distribution of f– values on IBP molecule. Fig. 5c presents the proposed IBP degradation pathway based on intermediates identification. Higher f– value of site on IBP molecule means more easily to lose an electron and be attacked by SO4·- and ·OH. The C3 (f– = 0.193) and C6 (f– = 0.196) show highest f– values, which are the most active site on IBP. It is consistent with the MS detection intermediates as the formation of products B and C due to the C—C bond cleavage caused by radical attacking (Fig. 5c). Moreover, ·OH addition pathway was also found for the formation of products D and E. Product F was further generated after E or C was attacked by ·OH. Finally, low molecular weight organic compounds and mineralization products (CO2, H2O and CO32-) formed after deep oxidation by the radicals. Total organic carbon (TOC) test indicated 25.2% of organic C was mineralized during IBP removal by h-Co(OH)2 activated PMS (Fig. S5 in Supporting information).

|
Download:
|
| Fig. 5. Chemical structure of IBP (a), natural population analysis (NPA) charges and Fukui index (f-) of IBP (b) and degradation pathway of IBP in the PMS activation system (c). | |
{kind=link}
This study developed an optimized solvothermal-hydrothermal method for preparation of hollow structure Co(OH)2, which could efficiently activate PMS for ibuprofen degradation. TEM and XRD confirmed the synthesized h-Co(OH)2 maintained microscale hollow spheres with thin nanosheets shell, and is assigned to α- Co(OH)2 crystalline phase. Compared with conventional p-Co (OH)2, the PMS activation activity of h-Co(OH)2 was significantly enhanced as well as the IBP removal efficiency. The rate constant (k1), interpreted by the pseudo-first order kinetic model, increased from 0.195 min-1 for p-Co(OH)2 to 0.428 min-1 for h-Co(OH)2. After PMS activation by h-Co(OH)2, the formed ·OH and SO4·- contributed to 6.3% and 75.7% to IBP degradation respectively, indicating the SO4·- played the dominant role. DFT calculation indicated that the sites of IBP molecule with high Fukui index (f-) was preferred to be attacked by the two produced electrophilic radicals. The hollow structure Co(OH)2 is a promising material for PMS activation, which shows great potential in efficient organic contaminants removal from water and wastewater.
AcknowledgmentsThis work was partially supported by the National Natural Science Foundation of China (Nos. 21906001 and 51721006). This work is supported by MOE Key Laboratory of Resources and Environmental Systems Optimization (NCEPU).
Appendix A. Supplementary dataSupplementary material related to this article can befound, inthe online version, at doi:https://doi.org/10.1016/j.cclet.2019.09.031.
| [1] |
T. aus der Beek, F.A. Weber, A. Bergmann, et al., Environ. Toxicol. Chem. 35 (2016) 823-835. DOI:10.1002/etc.3339 |
| [2] |
J. Wang, S. Wang, J. Environ. Manag. 182 (2016) 620-640. DOI:10.1016/j.jenvman.2016.07.049 |
| [3] |
V. Christen, S. Hickmann, B. Rechenberg, K. Fent, Aquat. Toxicol. 96 (2010) 167-181. DOI:10.1016/j.aquatox.2009.11.021 |
| [4] |
T.B. Veras, A. Luiz Ribeiro de Paiva, M.M.M.B. Duarte, D.C. Napoleão, J.J. da Silva Pereira Cabral, Chemosphere 222 (2019) 961-969. DOI:10.1016/j.chemosphere.2019.01.167 |
| [5] |
F. Méndez-Arriaga, S. Esplugas, J. Giménez, Water Res. 42 (2008) 585-594. DOI:10.1016/j.watres.2007.08.002 |
| [6] |
T. Takagi, C. Ramachandran, M. Bermejo, et al., Mol. Pharm. 3 (2006) 631-643. DOI:10.1021/mp0600182 |
| [7] |
J. Wang, B. He, D. Yan, X. Hu, Sci. Total Environ. 603- 604 (2017) 772-784. |
| [8] |
A. Białk-Bielinska, J. Kumirska, M. Borecka, et al., J. Pharm. Biomed. Anal. 121 (2016) 271-296. DOI:10.1016/j.jpba.2016.01.016 |
| [9] |
M. Jiménez-Salcedo, M. Monge, M.T. Tena, Chemosphere 215 (2019) 605-618. DOI:10.1016/j.chemosphere.2018.10.053 |
| [10] |
L. Lin, W. Jiang, M. Bechelany, et al., Chemosphere 220 (2019) 921-929. DOI:10.1016/j.chemosphere.2018.12.184 |
| [11] |
C. Aristizabal-Ciro, A.M. Botero-Coy, F.J. López, G.A.J.E.S. Peñuela, Environ. Sci. Pollut. Res. Int. 24 (2017) 7335-7347. DOI:10.1007/s11356-016-8253-1 |
| [12] |
J. Yan, J. Peng, L. Lai, et al., Environ. Sci. Technol. 52 (2018) 14302-14310. DOI:10.1021/acs.est.8b03340 |
| [13] |
H. Zhang, Q. Ji, L. Lai, G. Yao, B. Lai, Chin. Chem. Lett. 30 (2019) 1129-1132. DOI:10.1016/j.cclet.2019.01.025 |
| [14] |
J. Cao, L. Lai, B. Lai, et al., Chem. Eng. J. 364 (2019) 45-56. DOI:10.1016/j.cej.2019.01.113 |
| [15] |
Z. Wei, Y. Li, J. Wang, H. Li, Y. Wang, Chin. Chem. Lett. 29 (2018) 815-818. DOI:10.1016/j.cclet.2018.01.020 |
| [16] |
Y. Zhang, D. Zhang, X. Xu, B. Zhang, Chin. Chem. Lett. 29 (2018) 1350-1354. DOI:10.1016/j.cclet.2018.03.009 |
| [17] |
F. Ghanbari, M. Moradi, F. Gohari, J. Water Proc. Eng. 9 (2016) 22-28. DOI:10.1016/j.jwpe.2015.11.011 |
| [18] |
P. Devi, U. Das, A.K. Dalai, Sci. Total Environ. 571 (2016) 643-657. DOI:10.1016/j.scitotenv.2016.07.032 |
| [19] |
X. Lei, M. You, F. Pan, et al., Chin. Chem. Lett. (2019), doi:http://dx.doi.org/10.1016/j.cclet.2019.05.039.
|
| [20] |
A. Hussain, Y. Liu, T. Bin-aftab, D. Li, W. Sand, ChemNanoMat 5 (2019) 547-557. DOI:10.1002/cnma.201800677 |
| [21] |
G.P. Anipsitakis, D.D. Dionysiou, Environ. Sci. Technol. 37 (2003) 4790-4797. DOI:10.1021/es0263792 |
| [22] |
J. Zhao, Y.C. Zou, X.X. Zou, et al., Nanoscale 6 (2014) 7255-7262. DOI:10.1039/c4nr00002a |
| [23] |
J. Yang, H. Liu, W.N. Martens, R.L. Frost, J. Phy. Chem. C 114 (2010) 111-119. DOI:10.1021/jp908548f |
| [24] |
Z. Xu, Z. Chen, C. Joll, et al., Catal. Commun. 10 (2009) 1221-1225. DOI:10.1016/j.catcom.2009.01.021 |
| [25] |
X. Liu, R. Ma, Y. Bando, T. Sasaki, Adv. Funct. Mater. 24 (2014) 4292-4302. DOI:10.1002/adfm.201400193 |
| [26] |
G. Zhang, S. Zang, X. Wang, ACS Catal. 5 (2015) 941-947. DOI:10.1021/cs502002u |
| [27] |
J.T. Mehrabad, M. Aghazadeh, M.G. Maragheh, M.R. Ganjali, P. Norouzi, Mater. Lett. 184 (2016) 223-226. DOI:10.1016/j.matlet.2016.08.069 |
| [28] |
W. Li, H. Qi, B. Wang, et al., Microchim. Acta 185 (2018) 124. DOI:10.1007/s00604-017-2663-8 |
| [29] |
R. Luo, C. Liu, J. Li, et al., J. Mater. Chem. A 6 (2018) 3454-3461. DOI:10.1039/C7TA11052A |
| [30] |
W.D. Oh, Z. Dong, T.T. Lim, Appl. Catal. B 194 (2016) 169-201. DOI:10.1016/j.apcatb.2016.04.003 |
| [31] |
P. Hu, M. Long, Appl. Catal. B 181 (2016) 103-117. DOI:10.1016/j.apcatb.2015.07.024 |
| [32] |
M.J. Alowitz, M.M. Scherer, Environ. Sci. Technol. 36 (2002) 299-306. DOI:10.1021/es011000h |
| [33] |
Q. Chen, L. Chen, J. Qi, et al., Chin. Chem. Lett. 30 (2019) 1214-1218. DOI:10.1016/j.cclet.2019.03.002 |
| [34] |
H. Ji, Y. Zhu, J. Duan, W. Liu, D. Zhao, Chin. Chem. Lett. (2019), doi:http://dx.doi.org/10.1016/j.cclet.2019.06.004.
|
| [35] |
J. Wang, S. Wang, Chem. Eng. J. 334 (2018) 1502-1517. DOI:10.1016/j.cej.2017.11.059 |
| [36] |
F. Ghanbari, M. Moradi, Chem. Eng. J. 310 (2017) 41-62. DOI:10.1016/j.cej.2016.10.064 |
| [37] |
P. Shi, X. Dai, H. Zheng, et al., Chem. Eng. J. 240 (2014) 264-270. DOI:10.1016/j.cej.2013.11.089 |
| [38] |
F.J. Rivas, O. Gimeno, T. Borallho, Chem. Eng. J. 192 (2012) 326-333. DOI:10.1016/j.cej.2012.03.055 |
| [39] |
Y. Ren, L. Lin, J. Ma, et al., Appl. Catal. B 165 (2015) 572-578. DOI:10.1016/j.apcatb.2014.10.051 |
| [40] |
P. Neta, R.E. Huie, A.B. Ross, J. Phys. Chem. Ref. Data 17 (1988) 1027-1284. DOI:10.1063/1.555808 |
| [41] |
Y. Zhu, S. Chen, X. Quan, Y. Zhang, RSC Adv. 3 (2013) 520-525. DOI:10.1039/C2RA22039C |
| [42] |
Y.F. Huang, Y.H. Huang, J. Hazard. Mater. 162 (2009) 1211-1216. DOI:10.1016/j.jhazmat.2008.06.008 |
| [43] |
M. Abdul Nasir Khan, P. Kwame Klu, C. Wang, et al., Chem. Eng. J. 363 (2019) 234-246. DOI:10.1016/j.cej.2019.01.129 |
| [44] |
C. Guo, J. Xu, S. Wang, et al., Catal. Sci. Technol. 3 (2013) 1603-1611. DOI:10.1039/c3cy20811g |
| [45] |
Z.F. Huang, J.J. Zou, L. Pan, et al., Appl. Catal. B 147 (2014) 167-174. DOI:10.1016/j.apcatb.2013.08.038 |
| [46] |
X. Zhao, Z. Cai, T. Wang, et al., Appl. Catal. B 187 (2016) 134-143. DOI:10.1016/j.apcatb.2016.01.010 |
| [47] |
W. Liu, Y. Li, F. Liu, et al., Water Res. 151 (2019) 8-19. DOI:10.1016/j.watres.2018.11.084 |
| [48] |
E.T. Yun, J.H. Lee, J. Kim, H.D. Park, J. Lee, Environ. Sci. Technol. 52 (2018) 7032-7042. DOI:10.1021/acs.est.8b00959 |
| [49] |
F. de Vleeschouwer, V. van Speybroeck, M. Waroquier, P. Geerlings, F. de Proft, Org. Lett. 9 (2007) 2721-2724. DOI:10.1021/ol071038k |