 2019, Vol. 30
2019, Vol. 30 


b The Second Hospital of Jilin University, Changchun 130041, China;
c Key Laboratory for Molecular Enzymology and Engineering of Ministry of Education, School of Life Sciences, Jilin University, Changchun 130012, China
Glycosylation, the covalent addition of carbohydrates structures to amino acid on proteins, is most ubiquitous and structurally diverse forms of post-translational modification of eukaryotes [1]. Dynamic changes in protein glycosylation have been perceived to be closely relevant to various biological functions and pathological advances in many diseases. The majority of the discovered early diagnostic biomarkers and major therapeutic targets have been proved to be glycosylated proteins or peptides [2, 3]. Thereupon, it is vital important to develop glycoproteome identification techniques for in-depth understanding of glycoprotein functions and recognition of specific glycosylation patterns [4, 5]. Recently, mass spectrometry (MS) has been deemed as the powerful technology for glycoproteomics research on account of its fast analysis speed, less sample consumption, high throughput, and easy operation [6, 7]. However, the complexity of glycosylation site heterogeneity, the immanent low-abundance and ionization efficiency of glycopeptides, and other inferences in complex biological samples pose substantial challenges for MS-based strategies [8, 9]. Specific enrichment and isolation of glycoproteins/glycopeptides are therefore requisite for further MS analysis from complex samples [10, 11].
Up to now, multifarious strategies including hydrazide-mediated methods [12], lectin affinity chromatography [13], boronic acid-based covalent capture skills [14, 15], and hydrophilic interaction liquid chromatography (HILIC) [16-19] have been introduced to satisfy the demand for glycopeptides enrichment. Thereinto, HILIC manifests many merits such as excellent biocompatibility, good reproducibility, and unbiased adsorption towards diverse glycopeptides, which make it a promising approach to trap glycopeptides. To date, numerous HILIC nanomaterials based on various substrates such as sepharose [20], graphene oxide [21], monolith [2], and magnetic nanoparticles (MNPs) [22, 23] have been developed and acquired different enrichment performance in glycopeptides extraction. In particular, MNPs stand out due to their remarkable properties like rapid magnetic response, relatively large specific surface area and easy surface functionalization [24]. Benefiting from these distinctive properties, MNPs simplify the separation process and shorten the enrichment time [25-27], which endow them with wide applications in glycoproteomics research [28-30].
Various hydrophilic moieties have been grafted onto MNPs to prepare MNPs-based HILIC materials focusing on fast isolation and highly efficient capture for glycopeptides. For instance, Chen's group synthesized a series of maltose functionalized magnetic materials and applied to the isolation of glycopeptides from horseradish peroxidase (HRP) and immunoglobulin (IgG) digests [29]. Deng and co-workers prepared magnetic zwitterionic hydrophilic nanoparticles for glycopeptides enrichment with low detection limit (0.04 ng/μL), high selectivity (a mixture of HRP and BSA at the mass ratio of 1:50) and reproducibility (5 repeating cycles) [30]. However, cumbersome synthesis processes usually pose challenge to various hydrophilic moieties incorporation on MNPs. Thus, it is highly desirable to fabricate hydrophilic nanocomposites by facile synthesis steps for achieving superior enrichment specificity of glycopeptides.
Glutathione (GSH), a hydrophilic tripeptide composed of glutamic acid, cysteine and glycine, possesses superior biocompatibility and abundant polar groups such as carboxyl, amino, thiol and amide bonds, which exhibits great potential in glycopeptides enrichment [31-35]. Herein, GSH functionalized MNPs, designed as Fe3O4@Au-GSH, were synthesized by a simple two-step strategy. The as-synthesized composites possessed magnetic core of Fe3O4 to realize rapid separation. Massive gold nanoparticles were adhered to Fe3O4 core to not only avoid the aggregation of Fe3O4 NPs [36], but also offer ample active sites for further introduction of GSH with abundant polar groups via Au-SH interaction. The designed Fe3O4@Au-GSH was successfully applied to selectively enrich glycopeptides in complicated samples and showed superior performance.
The synthesis procedure [37] was represented in Fig. 1A. Generally, Fe3O4 NPs were firstly prepared with the classical ternary reaction system, i.e., FeCl3·6H2O, EG and NaAc. Next, Au NPs were prepared through the reduction of HAuCl4 aqueous solution by NaBH4 and adhered directly on the surface of Fe3O4 NPs by electrostatic adherence [38]. Finally, hydrophilic GSH was anchored onto Au NPs via Au-SH and the ultimate product was obtained. The as-prepared magnetic materials exhibited three superior merits. First, the existence of Fe3O4 NPs endowed the nanocomposites with super paramagnetic property. In contrast with other substrates, this feature made the separation process time-saving and convenient. Second, a high density attachment of Au NPs on Fe3O4 not only enhanced the biocompatibility of Fe3O4 but also contributed to the graft of more hydrophilic GSH, which enhanced the enrichment performance towards glycopeptides. More importantly, the whole synthesis procedure was facile and could be accomplished through a rapid two-step routine. The workflow of glycopeptides enrichment was depicted in Fig. 1B, which included four steps: incubation, washing, eluting and MALDI-TOF MS analysis.

|
Download:
|
| Fig. 1. (A) Synthesis procedure of Fe3O4@Au-GSH and (B) workflow of glycopeptides enrichment by Fe3O4@Au-GSH. | |
{kind=link}
The morphologies and dimensions of Fe3O4@Au and Fe3O4@AuGSH were investigated by TEM. The results shown in Fig. 2A indicated that Au NPs were successfully adhered on the surface of Fe3O4 and the as-prepared Fe3O4@Au had an average diameter of 189 nm (Fig. S1A in Supporting information). As displayed in Fig. 2B, after GSH was introduced into Fe3O4@Au NPs, the average diameter of the finally material increased about 30 nm (Fig. S1B in Supporting information), suggesting the successful synthesis of Fe3O4@Au-GSH.

|
Download:
|
| Fig. 2. TEM images of (A) Fe3O4@Au and (B) Fe3O4@Au-GSH. | |
{kind=link}
XRD patterns of the as-synthesis Fe3O4 and Fe3O4@Au-GSH were determined from 20° to 80° (2θ) and illustrated in Fig. S2 (Supporting information). In the case of Fe3O4 NPs, 6 diffraction peaks at 2θ = 30.5°, 35.6°, 43.3°, 53.7°, 57.3° and 63.1° could be observed in Fig. 2A, which were represented to (220), (311), (400), (422), (511) and (440) planes of Fe3O4, respectively (Fe3O4, JCPDS No. 82-1533) [39]. While for the Fe3O4@Au-GSH, except the characteristic peaks of Fe3O4, 4 additional peaks at 38.5°, 44.6°, 64.7°, and 77.8° corresponding to (111), (200), (220), and (311) planes of Au NPs also appeared (JCPDS No. 04-0784) [40]. It indicated that Au NPs do exist in the Fe3O4@Au-GSH nanocomposites and the crystalline structures of Fe3O4 were not destroyed by the subsequent synthesis process.
To further validate the successful attachment of GSH on Fe3O4@Au nanocomposites, FT-IR spectra of Fe3O4 NPs, GSH, and Fe3O4@Au-GSH were determined respectively and depicted in Fig. S3 (Supporting information). As shown in Fig. S3, the typical adsorption peak at 585 cm-1 was ascribed to Fe-O stretching vibration, while the peaks at 3445 and 1635 cm-1 were contributed to the remainder water in the prepared Fe3O4 NPs. The characteristic peak of GSH could be observed apparently at 2525 cm-1 belonged to S-H stretching and fingerprint region (the shaded area, from 1300 cm-1 to 400 cm-1) also appeared in the spectrum. Compared with unmodified Fe3O4 NPs, the peak of Fe-O stretching vibration in the functional Fe3O4@Au-GSH had a slight movement which was shift from 585 cm-1 to high wavenumbers of about 590 cm-1. This phenomenon could be explicated by the enhancement of the surface bond force according to a previous literature [41]. More importantly, the S-H stretching was disappeared due to the interaction of Au and S-H, which indicated that GSH was successfully modified onto the as-prepared nanocomposites. Furthermore, the wide and strong peak at 3420 cm-1 corresponded to the overlap of N-H and O-H stretching vibration, and 1642 and 1558 cm-1 were assigned to the characteristic absorption of the stretching vibration of the C = O and deformation vibration of N–H of amide group in the modified GSH, respectively. While the peak at 1398 cm-1 may be attributed to the bending vibration of O-H of carboxylic acid in GSH, and the peak at 668 cm-1 could owed to the vibration of C-S. The above spectrum information all revealed the successful synthesis of the assynthesized materials.
The thermal stability of Fe3O4@Au-GSH nanocomposites was indicated by thermal gravimetricanalyzer (TGA) with temperature ranged from 20 ℃ to 800 ℃. According to the TGA picture given in Fig. S4 (Supporting information), the slight weight loss before 200 ℃ could be ascribed to the removal moisture or organic small molecules on the materials. Compared with the TGA curves of Fe3O4@Au, Fe3O4@Au-GSH had a mass loss of 4.6%, which was contributed to the departure of GSH. Finally, the TGA curves basically levelled off at 800 ℃, and the content of GSH grafted onto the Fe3O4@Au-GSH was estimated to be about 149.7 μmol/g from the TGA data analysis [28].
In HILIC mode, the content of ACN was of great concern for the adsorption efficiency of glycopeptides. The difference between glycopeptides and non-glycopeptides could be enlarged by adjusting the proportion of ACN in the loading buffer [42]. Therefore, we investigated four loading solutions with different ACN concentrations for glycopeptides capture in HRP tryptic digest. Five high-signal intensity glycopeptides were chose as markers to estimate the enrichment performance (Fig. S5A in Supporting information). It was found that glycopeptides were well retained with high abundance when the ratio of ACN reached 95%, while the higher or lower ACN concentration would lead to the decrease of glycopeptides signals. Thereupon, 95% ACN was applied as the optimal proportion in loading buffer. Besides, we further investigated the effect of acidity on enrichment ability. Results indicated that loading buffers with 3% TFA was the best for glycopeptides capture (Fig. S5B in Supporting information). Analogously, the influence of ACN and acidity on eluting buffer was also explored (Figs. S5C and S5D in Supporting information). Eventually, we chose the solution with a composition of 95% ACN/ 3% TFA (v/v) as the optimal loading buffer, 70% ACN/1% TFA (v/v) as the most appropriate eluting buffer and applied them to subsequent analysis.
To assess the enrichment performance of as-prepared HILIC materials for glycopeptides, Fe3O4@Au-GSH was exploited to capture glycopeptides from HRP (a standard glycoprotein) tryptic digestion under the optimal conditions. As we can see from Fig. 3A, non-glycopeptides had strong signal interference so that the signal intensity of glycopeptides were severely suppressed, however, it could be found that 22 glycopeptides with high signal abundance distinctly appeared and non-glycopeptides were efficiently eliminated after enrichment with Fe3O4@Au-GSH (Fig. 3B). In addition, IgG digests containing different kinds of glycans were investigated (Fig. S6 in Supporting information). Before enrichment, only 6 peaks could be detected. After enrichment with Fe3O4@Au-GSH, 23 glycopeptides were detected with high intensity, which demonstrated that this method showed unbiased enrichment ability toward glycopeptides with different glycan types. The detailed information about identified glycopeptides from HRP and IgG digests were displayed in Tables S1 and S2 (Supporting information), respectively. These acquired results might contributed to the hydrogen bonding between the polar groups of GSH and glycans of glycopeptides [42], which demonstrated that the superior hydrophilicity of GSH largely facilitated the as-prepared HILIC materials for the specific adsorption of glycopeptides.

|
Download:
|
Fig. 3. MALDI-TOF mass spectra of HRP tryptic digests (A) before and after enrichment by Fe3O4@Au-GSH with different concentrations: (B) 23 pmol/μL; (C) 50 fmol/μL; (D) 10 fmol/μL. Mass spectrometric peaks of glycopeptides were marked with  |
|
{kind=link}
In consideration of the extremely low abundance of glycopeptides in complex real bio-samples, the sensitivity was an essential index for glycopeptides enrichment. For this purpose, various concentrations of HRP digests incubated with Fe3O4@Au-GSH were investigated. As shown in Fig. 3C, 17 peaks originated from the target analyte were clearly observed when the concentration of HRP digests were diluted to 50 fmol/μL. Even if the concentration of HRP digest decreased to 10 fmol/μL (Fig. 3D), 10 peaks of glycopeptides could be still detected (for which S/N signal over 10). The above consequence manifested that Fe3O4@Au-GSH had the low detection limit at fmol level for glycopeptides identification and possessed the potential to detect glycopeptides signal peaks in complicated samples.
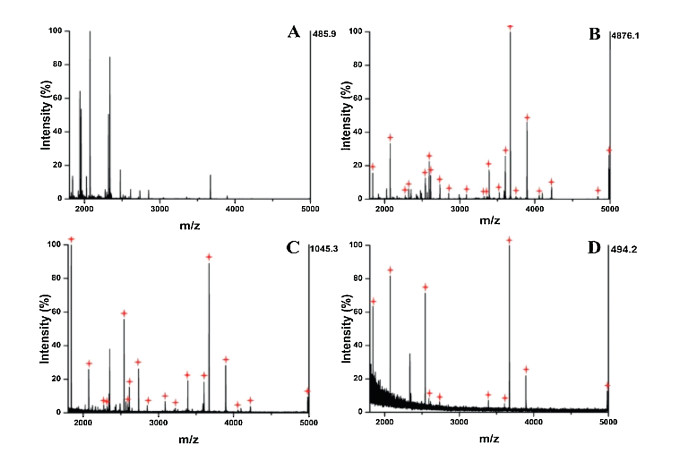
The specificity of glycopeptides capture by Fe3O4@Au-GSH were also evaluated by the mimic samples containing tryptic digests of HRP and BSA (non-glycoprotein) at different mass ratios (1:30, 1:50, 1:100) and the results were shown in Fig. 4. The mass spectrum of the mimic complex samples indicated that only nonglycopeptides could be observed, whereas 3 peaks were detected for glycopeptides with a mass ratio of 1:50 without treatment (Fig. 4A). After enrichment by Fe3O4@Au-GSH, 14 and 13 glycopeptides signals were identified when the mass ratio was 1:30 (Fig. 4B) and 1:50 (Fig. 4C), respectively. Even when the ratio enhanced to 1:100, 11 peaks could still detected with moderate intensity (Fig. 4D). Based on the satisfactory results mentioned above, it is expected that the proposed Fe3O4@Au-GSH with low detection limit and high selectivity could be applied to more complex matrix.

|
Download:
|
|
Fig. 4. MALDI-TOF mass spectra of mixtures of HRP and BSA tryptic digests (A) before and after enrichment by Fe3O4@Au-GSH with different mass ratios: (B) 1:30; (C) 1:50; (D) 1:100. Mass spectrometric peaks of glycopeptdes were marked with |
|
{kind=link}
The repeatability of Fe3O4@Au-GSH was investigated by enriching glycopeptides from HRP digests in consecutive times. After each run of enrichment process, Fe3O4@Au-GSH was purged with the eluting buffer several times to remove the residue adhered on the surface of as-fabricated materials. The results were shown in Fig. S7 (Supporting information), glycopeptides signals were clearly detected in each cycle and there were no obvious difference of glycopeptides peaks between the first time and the fifth times run, demonstrating that Fe3O4@Au-GSH owed glorious reusability and stability for glycopeptides capture. What is more, the comparison study about the enrichment performance between the developed strategy and other reported hydrophilic methods demonstrated that the present approach provided comparable sensitivity, selectivity, and reagent dosage [18, 23, 28, 30].
The enrichment capacity of glycopeptides by Fe3O4@Au-GSH was further evaluated towards heterogeneous complicated samples. Human serum was selected as practical complex matrix [43, 44]. As shown in Fig. S8 (Supporting information), the nonglycopeptides peaks in human serum dominated the mass spectrum without any treatment. While after incubated with Fe3O4@Au-GSH nanocomposites, 22 peaks originated from human serum were discriminated. Table S3 (Supporting information) listed the detailed information about identified glycopeptides from human serum. These results suggested that the hydrophilic Fe3O4@Au-GSH had great potential to be applied to glycopeptides identification from complicated samples.
In summary, we proposed a facile strategy to synthesize the hydrophilic glutathione modified magnetic nanoparticles and subsequently applied it to the capture of glycopeptides from complicated samples. The as-fabricated materials combined merits such as superparamagnetism from Fe3O4 NPs, superior biocompatibility and glorious hydrophilicity from Au NPs and GSH, exhibiting high selectivity and great potential in glycopeptides enrichment. Hydrogen bonding and multivalent interaction endowed the materials with good enrichment efficiency toward glycopeptides. We expected that this work could be conducive to the construction of hydrophilic materials and also pave a way for the capture of target analyte from complicated matrices.
AcknowledgmentsThis project was supported by Jilin Provincial Science & Technology Department (No. 20190201079JC), the Fundamental Research Funds for the Central Universities, JLU and Open Project of State Key Laboratory of Supramolecular Structure and Materials, Jilin University, China (No. sklssm2019020).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.06.046.
| [1] |
J. Stadlmann, J. Taubenschmid, D. Wenzel, et al., Nature 549 (2017) 538-542. DOI:10.1038/nature24015 |
| [2] |
H. Zheng, X. Li, Q. Jia, ACS Appl. Mater. Inter. 10 (2018) 5909-5917. DOI:10.1021/acsami.7b18999 |
| [3] |
S. Sun, P. Shah, S.T. Eshghi, et al., Nat. Biotechnol. 34 (2016) 84-88. DOI:10.1038/nbt.3403 |
| [4] |
X. Wang, Y. Liu, F. Li, Z. Li, Chin. Chem. Lett. 28 (2017) 1018-1026. DOI:10.1016/j.cclet.2017.02.001 |
| [5] |
F. Ding, Z. Chu, Q. Zhang, H. Liu, W. Zhang, Anal. Chim. Acta 1057 (2019) 145-151. |
| [6] |
L. Dang, J. Shen, T. Zhao, et al., Anal. Chem. 91 (2019) 5478-5482. DOI:10.1021/acs.analchem.8b05639 |
| [7] |
C. Qi, H. Jiang, J. Xiong, B. Yuan, Y. Feng, Chin. Chem. Lett. 30 (2019) 553-557. DOI:10.1016/j.cclet.2018.11.029 |
| [8] |
Y. Zhang, H. Jing, T. Wen, et al., Talanta 191 (2019) 509-518. DOI:10.1016/j.talanta.2018.09.016 |
| [9] |
R. Xing, Y. Wen, H. He, Z. Guo, Z. Liu, TrAC-Trends. Anal. Chem. 110 (2019) 417-428. DOI:10.1016/j.trac.2018.11.033 |
| [10] |
P. Wang, H. Zhu, J. Liu, et al., Chem. Eng. J. 358 (2019) 143-152. DOI:10.1016/j.cej.2018.09.168 |
| [11] |
B. Luo, Q. Chen, J. He, et al., ACS Sustain. Chem. Eng. 7 (2019) 6043-6052. DOI:10.1021/acssuschemeng.8b06171 |
| [12] |
L. Liu, M. Yu, Y. Zhang, C. Wang, H. Lu, ACS Appl. Mater. Interfaces 6 (2014) 7823-7832. DOI:10.1021/am501110e |
| [13] |
B. Zhang, R. Yu, Y. Yu, et al., Analyst 143 (2018) 5090-5093. DOI:10.1039/C8AN01258J |
| [14] |
H. Xiao, W. Chen, J.M. Smeekens, R. Wu, Nat. Commun. 9 (2018) 1692. DOI:10.1038/s41467-018-04081-3 |
| [15] |
C. Zhou, X. Chen, Z. Du, et al., J. Chromatogr. A 1498 (2017) 90-98. DOI:10.1016/j.chroma.2017.01.049 |
| [16] |
X. He, X. Liang, X. Chen, et al., Anal. Chem. 89 (2017) 9712-9721. DOI:10.1021/acs.analchem.7b01283 |
| [17] |
W. Ma, L. Xu, X. Li, et al., ACS Appl. Mater. Inter. 9 (2017) 19562-19568. DOI:10.1021/acsami.7b02853 |
| [18] |
L. Zhang, S. Ma, Y. Chen, et al., Anal. Chem. 91 (2019) 2985-2993. DOI:10.1021/acs.analchem.8b05215 |
| [19] |
J. Peng, Y. Hu, H. Zhang, et al., Anal. Chem. 91 (2019) 4852-4859. DOI:10.1021/acs.analchem.9b00542 |
| [20] |
H. Zhu, X. Li, J. Qu, et al., Anal. Bioanal. Chem. 409 (2017) 511-518. DOI:10.1007/s00216-016-9937-6 |
| [21] |
Y. Chen, Q. Sheng, Y. Hong, M. Lan, Anal. Chem. 91 (2019) 4047-4054. DOI:10.1021/acs.analchem.8b05578 |
| [22] |
J. Li, F. Wang, J. Liu, et al., Chem. Commun. 51 (2015) 4093-4096. DOI:10.1039/C5CC00187K |
| [23] |
N. Sun, J. Wang, J. Yao, C. Deng, Anal. Chem. 89 (2017) 1764-1771. DOI:10.1021/acs.analchem.6b04054 |
| [24] |
J. Wang, J. Li, Y. Wang, et al., ACS Appl. Mater. Inter. 8 (2016) 27482-27489. DOI:10.1021/acsami.6b08218 |
| [25] |
Y. Dai, L. Niu, J. Zou, et al., Chin. Chem. Lett. 29 (2018) 887-891. DOI:10.1016/j.cclet.2017.11.029 |
| [26] |
J. Li, X. Long, H. Yin, J. Qiao, H. Lian, J. Sep. Sci. 39 (2016) 2562-2572. DOI:10.1002/jssc.201600231 |
| [27] |
Z. Chen, B. Chen, M. He, H. Wang, B. Hu, Microchim. Acta 186 (2019) 107. DOI:10.1007/s00604-018-3139-1 |
| [28] |
C. Bi, Y. Liang, L. Shen, et al., ACS Omega 3 (2018) 1572-1580. DOI:10.1021/acsomega.7b01788 |
| [29] |
C. Bi, Y. Zhao, L. Shen, et al., ACS Appl. Mater. Inter. 7 (2015) 24670-24678. DOI:10.1021/acsami.5b06991 |
| [30] |
R. Wu, Y. Xie, C. Deng, Talanta 160 (2016) 461-469. DOI:10.1016/j.talanta.2016.07.045 |
| [31] |
D. Fu, Y. Liu, A. Shen, et al., Anal. Bioanal. Chem. 411 (2019) 4131-4140. DOI:10.1007/s00216-019-01661-0 |
| [32] |
Q. Liu, C. Deng, N. Sun, Nanoscale 10 (2018) 12149-12155. DOI:10.1039/C8NR03174F |
| [33] |
Y. Ma, L. Wang, Y. Zhou, X. Zhang, Nanoscale 11 (2019) 5526-5534. DOI:10.1039/C9NR00392D |
| [34] |
N. Sun, Z. Wang, J. Wang, et al., J. Chromatogr. A 1595 (2019) 1-10. DOI:10.1016/j.chroma.2019.02.039 |
| [35] |
X. Feng, C. Deng, M. Gao, X. Zhang, Anal. Bioanal. Chem. 410 (2018) 989-998. DOI:10.1007/s00216-017-0602-5 |
| [36] |
W. Zhang, Y. Zhang, Q. Jiang, et al., Anal. Chem. 88 (2016) 10523-10532. DOI:10.1021/acs.analchem.6b02583 |
| [37] |
D. Jiang, X. Li, J. Ma, Q. Jia, Talanta 180 (2018) 368-375. DOI:10.1016/j.talanta.2017.12.048 |
| [38] |
J. Ren, S. Shen, Z. Pang, et al., Chem. Commun. 47 (2011) 11692-11694. DOI:10.1039/c1cc15528h |
| [39] |
S. Karamipour, M. Sadjadi, N. Farhadyar, Spectrochim. Acta PartA:Mol. Biomol. Spectroscopy 148 (2015) 146-155. DOI:10.1016/j.saa.2015.03.078 |
| [40] |
H. Zhang, X. Zhong, J. Xu, H. Chen, Langmuir 24 (2008) 13748-13752. DOI:10.1021/la8028935 |
| [41] |
M. Ma, Y. Zhang, W. Yu, et al., Colloids Surf. A:Physicochem. Eng. Aspects 212 (2003) 219-226. DOI:10.1016/S0927-7757(02)00305-9 |
| [42] |
S. Ma, L. Zhang, S. Wang, et al., Anal. Chim. Acta 1058 (2019) 97-106. DOI:10.1016/j.aca.2019.01.011 |
| [43] |
K. Sparbier, S. Koch, I. Kessler, T. Wenzel, M. Kostrzewa, J. Biomol. Tech. 16 (2005) 407-413. |
| [44] |
S. Wang, J. Ye, Z. Bie, Z. Liu, Chem. Sci. 5 (2014) 1135-1140. DOI:10.1039/c3sc52986j |