 2019, Vol. 30
2019, Vol. 30 


b Sino-German Centre for Water and Health Research, Sichuan University, Chengdu 610065, China;
c Institute of Environmental Engineering, RWTH Aachen University, Aachen 52072, Germany
In the recent decades, the increasingly severe water pollutions caused by various organic pollutants from industrial production, agricultural application, breeding industry, and anthropogenic discharges have attracted more and more attentions [1]. Although many conventional physicochemical methods (e.g., adsorption [2], coagulation [3], and Fe-C micro-electrolysis [4-6]) have been used to eliminate these organic pollutants from the contaminated water, the treatment efficiency is usually not satisfactory. Meanwhile, some unavoidable technical drawbacks including catalyst regen-eration difficulty, low treatment efficiency, and catalyst passiv-ation in the above methods limit their practical applications.
Advanced oxidation processes (AOPs) have been considered as a promising technology for the degradation of refractory, non-biodegradable, and toxic compounds. AOPs mainly depended on the generated free radicals to decompose organic pollutants [7-14]. Especially, hydroxyl radical (HO·, Eθ=2.8 V) is an effective strong oxidant. The oxidation capacity of hydroxyl radical is second only to fluorine [15], and the reaction rate between HO· and organic molecules usually reaches 106-109 mol -1 s-1 [16]. Generally, hydroxyl radical was mainly from the decomposition of H2O2 and O3 in the presence of activators (e.g., Co2+, Cu2+, and Fe2+). Recently, sulfate radical (SO4·-) based AOPs have obtained more and more attentions [17-19]. The sulfate radical has a similar or even higher redox potential (2.5–3.1 V) than hydroxyl radical. Moreover, sulfate radical processes a longer half-time (t1/2 = 30– 40 μs) than hydroxyl radical (t1/2 < 1 μs), which ensures sulfate radical to attack the targeted pollutant effectively [20]. Persulfate (PS) and peroxymonosulfate (PMS) are strong oxidants. However, the reaction rates between the above two oxidants with the organics were low. Normally, sulfate radical could be generated through various PS and PMS activation processes.
Electrochemical advanced oxidation processes (EAOPs) have been regarded as one of the most potential advanced oxidation processes, which have some special advantages such as environ-mental friendliness, strong catalytic ability, no secondary pollu-tion, low-volume application, mild operating condition, and easy operation [21, 22]. In particular, the EAOPs for the degradation of organic compounds could be used as the pre-treatment technology before the bio-treatment process [22]. In addition, the report shows that EAOPs could efficiently mineralize various azo dyes, pesticides, and some small acids [23]. Especially, electro-Fenton reaction was the most known EAOPs involving the Fenton process. It includes the generation of H2O2 on the cathode through the aerated air/oxygen. The added Fe2+ further reacted with H2O2 to form hydroxyl radical. The reduction of Fe3+ to Fe2+ accelerate the continue Fenton process and the generation of free radical. Due to the superiority of electrochemical Fenton system, the combined technologies of electrochemical advanced oxidation with oxidants such as PS, PMS, and O3 are proposed.
Herein, the objectives of the mini-review are to provide a comprehensive summary of the technology of electrochemical advanced oxidation processes combined with different oxidants (e.g., ozone, persulfate, and peroxymonosulfate) for the degrada-tion of organic compounds (Fig. 1). Moreover, the article reviews the reaction mechanisms of pollutants degradation in the coupling systems.

|
Download:
|
| Fig. 1. Scheme of the (photo)electro-oxidants system used for organic compounds degradation. | |
2. EAOPs-PS systems
The studies found that electro-activated persulfate system is an effective manner for the removal of various organic pollutants [24-26]. As we all know, sulfate radical would be generated through the electro-activated persulfate oxidation. The degradation of pollut-ant in electro-activated persulfate process was dependent on the sulfate radical formed by cathodic electron transfer (Eq. (1)) [26]. As reported, electro-activated persulfate process has been successfully applied to eliminate the organic compounds, such as sulfamethoxazole [27], hexachlorocyclohexanes [28], disperse blue 3 [29], tetracycline hydrochloride [30], and carbamazepine [25] in the aqueous solution. Furthermore, Chen et al. also studied the mineralization efficiency of dinitrotoluenes (DNTs) [24] and aniline [26, 31], respectively. It shows that DNTs could be almost degraded under the optimal conditions of electrode potential of 6 V, the temperature of 318 K, N2 rate of 150 mL/min, pH of 0.5 and persulfate anion concentration of 1.7 wt%, and the mineralization efficiency was 79% after 8 h [24]. What is more, Chen et al [31]. studied the degradation of aniline in the aqueous solution by the electro-activated persulfate system enhanced with ultrasound to accelerate the mass transfer and the generation of free radicals, which shows that the sonoelectro-persulfate system could obviously improve the total organic carbon (TOC) removal.
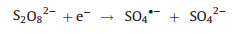
|
(1) |
Besides, some authors presented the homogeneous electro-chemical advanced oxidation processes in the presence of PS with the addition of homogeneous catalyst for different organic compounds degradation (Table 1). In the homogeneous EAOPs-PS systems, the main electrodes commonly consist of anode materials such as mixed metal oxides (e.g., Ti/RuO2-IrO2), iron sheet, carbon felt, and cathode with titanium, stainless steel, iron sheet [32, 33]. In EAOPs-PS coupling processes, the homogeneous catalyst (mostly Fe2+ and Fe3+) was added into the electro/ persulfate system. The reaction mechanism was as follows: on one hand, sulfate radical would be generated on the cathode through electron transfer (Eq. (1)). On the other hand, the added Fe2+ activator could activate PS to form sulfate radical, and then Fe3+ formed by Fe2+ oxidation can be further reduced at the cathode to form Fe2+ (Eqs. (2) and (3)). Especially, when the anode material was iron material [34, 35], Fe2+ was derived from the surface of iron anode (Eq. (4)), and then Fe2+ triggers the PS activation process.
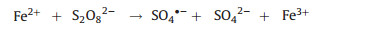
|
(2) |

|
(3) |

|
(4) |
|
|
Table 1 The degradation of organic compounds in the EAOPs-PS systems. |

However, homogeneous electrochemical advanced oxidation processes have difficulty in catalyst recovery, and the cost of catalyst separation is high. Thus, the solid catalyst in the EAOPs-PS systems has been an alternative of Fe2+/Fe3+ ions, and the interest of the study about heterogeneous electrochemical advanced oxidation processes have been the focus. Generally, the use of the solid catalyst includes nano-Fe@NdFeB/AC [43], natural maghemite (NM) [44], MnO2 [45], Fe3O4 [52], Fe-Cu/SBA-15 [46] (Table 1), which enable the lower dissolved metal ions and further reduce secondary pollution. The degradation mechanism of the organic pollutants was as follows: in addition to sulfate radicals produced by electron transfer on the cathode, the PS catalyst activation process could also occur on the surface of catalyst (single-metal oxide and mixed-metal oxide).
(ⅰ) Single-metal oxide catalysts: Zhang and his colleges investigat-ed the activity performance of Fe3O4 [47], MnO2 [45], maghemite [44], goethite [48], and Fe/SBA-15 [50] in the EAOPs-PS system, the mechanisms of PS activation by heterogeneous single-metal oxide catalysts can be expressed as follows (taking MnO2 for example):
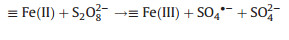
|
(5) |
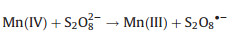
|
(6) |

|
(7) |
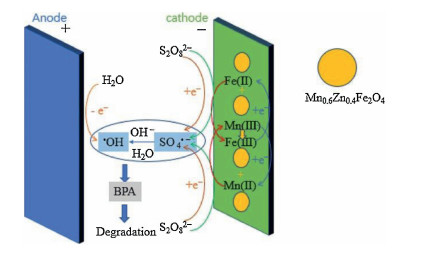
(ⅱ) Mixed-metal oxide catalysts: the performance of mixed-metal oxide such as MFe2O4 (e.g., Mn0.6Zn0.4Fe2O4 [49], CuFe2O4 [53]), and Fe-M oxides (e.g., Fe-Co/SBA-15 [51] and Fe-Cu/SBA-15 [46]) (M = Mn, Zn, Cu, Co, and etc.) have been studied by many researchers. Taking Mn0.6Zn0.4Fe2O4 as an example, the reaction mechanism of common magnetic Mn0.6Zn0.4Fe2O4 activation for PS was dependent on the redox pairs of Mn(Ⅲ)/ Mn(Ⅱ) and Fe(Ⅲ)/Fe(Ⅱ), which could be described by Eqs. (5, 8–11) (Fig. 2) [49]:
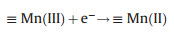
|
(8) |
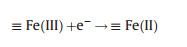
|
(9) |
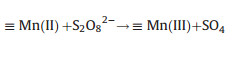
|
(10) |
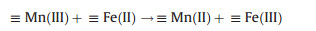
|
(11) |

|
Download:
|
| Fig. 2. Schematic diagram of the mechanism of the electro/Mn0.6Zn0.4Fe2O4/PDS process. Copied with permission [49]. Copyright 2018, Elsevier. | |
For the Fe-M oxides, the activation reaction was similar to the PS activation by MFe2O4 via the valence variation of Fe and M. For example, the activation mechanism of PS by Fe-Co/SBA-15 was based on the Fe(Ⅱ) and Co(Ⅱ) generated on cathode and the further PS activation by Fe(Ⅱ) and Co(Ⅱ), which could be expressed through the Eqs. (5), (9), (12) and (13) [51]:
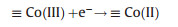
|
(12) |
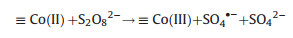
|
(13) |
Recently, the homogeneous catalysts Fe3+ [54, 55] and Fe2+ [56, 57] have been added into the EAOPs-PMS system. Generally, the degradation reactions in the homogeneous EAOPs-PMS could be summarized as follows: Fe2+ was generated from Fe3+ on the cathode (Eq. (3)), and then the generated Fe2+ further activates PMS to form sulfate radicals (Eq. (14)). Meanwhile, the sulfate radical also could be formed through electron transfer on the cathode (Eq. (15)), accompanying hydroxyl radical produced on the anode (Eq. (16)).
In addition, heterogeneous EAOPs-PMS systems have been investigated using hydronium jarosite [58], Co3O4 [59], activated carbon [60], iron-based minerals [61], and ZVI [62] as the heterogeneous catalyst. For instance, over 80% of TOC removal was obtained by EC/pyrite/PMS system degrading 1-butyl-1-methylpyrrolidinium chloride (1.85 mmol/L) with PMS of 10 mmol/L, current of 150 mA, and reaction time of 300 min with a low electrical energy consumption of 5.45 kWh m -3 order -1 [61]. Li et al. [60] found that 89.8% of AO7 (100 mg/L) removal was achieved during 60 min treatment process in the electro/GAC/PMS system under the optimal conditions of PMS concentration of 5 mmol/L, current density of 8 mA/cm2, GAC dosage of 0.5 g/L, Na2SO4 dosage of 50 mmol/L, and pH of 5. Heterogeneous EAOPs-PMS system follows a similar reaction mechanism as the heterogeneous EAOPs-PS system, and the improved pollutant degradation efficiency was ascribed to the synergistic effect in the reaction process (Fig. 3). Compared with the homogeneous EAOPs-PMS system, the heterogeneous EAOPs-PMS could effectively reduce the leaching of metal ions, decrease the secondary pollution, and facilitate the recycling of the catalyst.
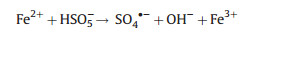
|
(14) |

|
(15) |
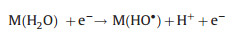
|
(16) |

|
Download:
|
| Fig. 3. Possible mechanism of AO7 elimination in the MFC/HJ/PMS system. Copied with permission [58].Copyright 2017, Elsevier. | |
4. EAOPs-peroxone systems
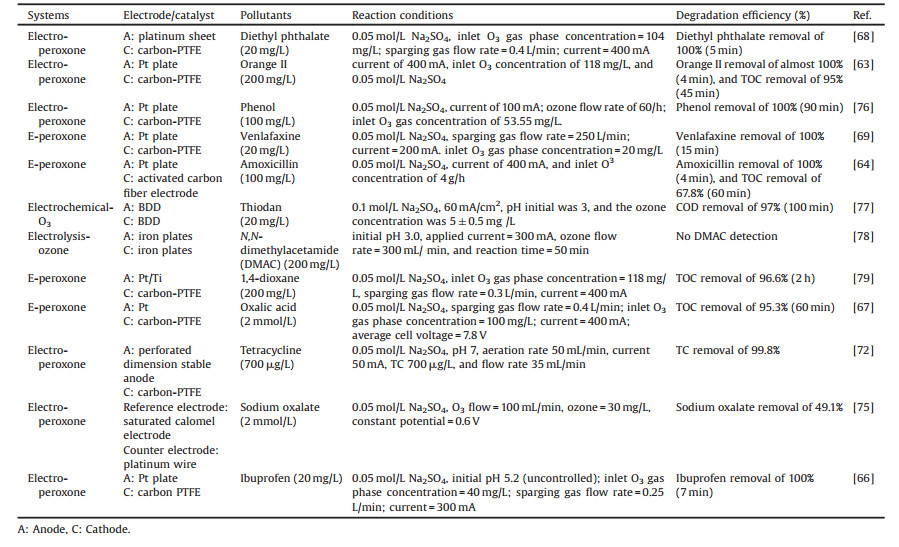
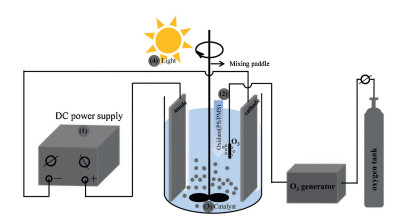
Electro-peroxone system is one of important EAOPs coupling technologies. The recent studies about the EAOPs-peroxone systems used for various organic compounds (e.g., orange II [63], amoxicillin [64], rhodamine B [65], ibuprofen [66], oxalic acid [67], and diethyl phthalate [68]) degradation are presented in Table 2. As shown in Fig. 4, in the electro-peroxone systems, the mixed gas (O3 and O2) from the ozone generator was purged into the reaction solution as the desired concentration. The O2 in the sparged gas would be converted into H2O2 on the cathode (Eq. (17)) [69, 70]. The conjugated based HO2- from Eq. (18) further reacts with O3 in the solution to produce HO· (Eqs. (19)–(22)) [67]. Differ from the previously recognized Eq. (23), recent reports indicated that the HO· formation efficiency was half of that obtained in Eq. (23) for the reason that HO5- would decompose into O2 and OH- (Eq. (24)) [71]. Except for the above HO· source, the O3 in sparged gas can also be reduced into HO· through the reaction (25) [72]. In general, the generation of H2O2 on the cathode is critical to drive the EAOPs-peroxone system, which is essential for the improvement of the pollutant elimination [73, 74].
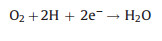
|
(17) |
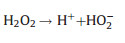
|
(18) |

|
(19) |
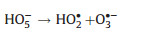
|
(20) |

|
(21) |
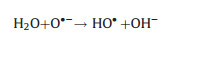
|
(22) |
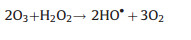
|
(23) |
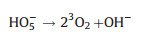
|
(24) |
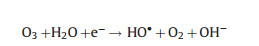
|
(25) |
|
|
Table 2 The degradation of organic compounds in the EAOPs-peroxone systems. |

|
Download:
|
| Fig. 4. The synergistic effect in the electroperoxone process using a three electrode system. Copied with permission [75].Copyright 2017, Elsevier. | |
Cathode material is an important parameter related to the degradation and mineralization efficiency of pollutants for the reason that it can significantly affect the formation of hydrogen peroxide and hydroxyl radicals in the EAOPs system. Hou et al. [68] studied the effects of cathodes materials on the diethyl phthalate (DEP) degradation efficiency in the EAOPs-peroxone system, which shows that the mineralization of DEP in EAOPs- peroxone system with carbon materials as a cathode was significantly higher than that with stainless steel as a cathode.
In addition, the mineralization efficiencies with the different carbon-based cathode materials were the following order: carbon felt < RVC < carbon-PTFE. Wang et al. [67] also found a similar result that the carbon-PTFE cathode material is beneficial for the production of H2O2, significantly accelerating the subsequent reactions about the production of free radical. What is more, the role of anode material on the oxalic acid mineralization in the O3-electrolysis system was reported by Wang et al. [67]. In their studies, when the anode material was changed from Pt to boron-doped diamond (BDD) while keeping other reaction conditions consistent, TOC removal improved from 70.2% to 97.6% at 2 h, which was attributed that the BDD anode is declined to produce HO in the presence of electrolysis for the wastewater treatment, while Pt anode is not. The above results suggested that the type of anode electrode could obviously affect the TOC removal when electrolysis coupled to the O3 oxidation.
Differ from the electro-peroxone system, Xiong and his co-workers [78] reported the N, N-dimethylacetamide (DMAC) degra-dation in the electrolysis-ozone process by using two iron plates with the same size as anode and cathode, respectively. In this electrolysis-ozone process, the iron anode would be as the sacrificial anode to release the highly reactive Fe2+ as the homogeneous catalyst to trigger Fenton-like reaction. The reaction mechanism was explained that the released Fe2+ from anode catalyzed the decomposition of ozone to generate HO· for the DMAC degradation (Eqs. (26) and (27)). What is more, in this electrolysis-ozone with iron plate as the cathode, the H2O2 could be generated indirectly by the ozone decomposition (Eqs. (28) and (29)), the generated H2O2 would involve in the Fenton-like and peroxone reaction with the nascent Fe2+ and O3, respectively. The formed Fe3+ from Fe2+ oxidation would precipitate by the form of Fe(OH)3 in the system and removal DMAC by flocculation. In conclusion, the contribution of catalytic ozonation, Fenton-like, peroxone, direct ozone oxidation, and coagulation for DMAC removal were about 56.3%, 31.6%, 7.7%, 3.0% and 1.4%, respectively.
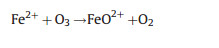
|
(26) |
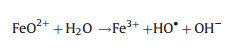
|
(27) |
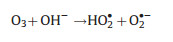
|
(28) |
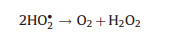
|
(29) |
Photoelectrocatalytic process is an excellent technology for organics degradation [80]. However, there is not much research on the photoelectro-oxidant coupling process, and some studies about photoelectron-oxidant system involving the oxidant (e.g., PS, PMS, and O3), UV irradiation, electrolysis and/or catalyst for organic pollutants degradation are shown in Table 3. The synergistic effect among the coupling systems could be highlighted due to the interaction of the individual process under the optimal conditions, accelerating the production of free radicals to attack the target pollutants. Bensalah et al. [81] studied the degradation of 4-nitrophenol (NP) by the photoelectro-peroxone (PEP) processes with a boron-doped diamond (BDD) anode. The H2O2 would generate through the reduction of O2 on carbon felt cathode, and then the generated H2O2 decomposed through UV irradiation and reacted with O3 to produce HO·.
|
|
Table 3 The degradation of organic compounds in the photoelectro-oxidant systems. |

In addition, Shen et al. [83] studied the 1, 4-dioxane degradation (200 mg/L) by combining the electrolysis, UV, and O3 under the optimal conditions, which shows that the synergistic factor of the coupling system was far higher than other control systems. The 1, 4-dioxane degradation was also discussed by the photoelectro-peroxone process [83]. The differences in cathode materials lead to differences in reaction mechanisms. Since H2O2 cannot be directly generated on cathode electrodes that are not carbon-based materials, the H2O2 is generated from reaction (30). What is more, the type of electrolyte has a significant impact on the degradation and mineralization of pollutants. Jaafarzadeh et al. [82] found the degradation efficiency of 2, 4-dichlorophenoxy-acetic acid significantly inhibited when sodium sulfate electrolyte was replaced by sodium chloride, sodium nitrate, sodium bicarbonate, and sodium dihydrogen phosphate, respectively. The result was ascribed that chloride ion would transfer to active chlorine species (Cl2, HClO and ClO-) in the presence of electrolysis, the formed HClO and ClO- would react with H2O2, resulting in a decrease in hydrogen peroxide concentration [83, 85]. Furthermore, it is well known that NO3-, HCO3- and H2PO4- are three anions scavengers of free radicals. However, the inhibition degree of 2, 4-dichlorophenoxyacetic acid degradation with NO3- as electrolyte is lower than that with Cl-, HCO3-, or H2PO4- for the reason that photolysis of nitrate and subsequent reactions can produce partial hydroxyl radicals. Thus, the SO42- was selected as the superior electrolyte due to its stability in the electrochemical process and the ability of reducing the energy consumption [82].
On the basis of the above-mentioned process, the researchers tried to add additional catalyst in the photoelectron-peroxone system to improve the synergistic effect and promote pollutant degradation. Photoelectro-peroxone/ZVI process with graphite felt as a cathode was used for the organic pollutant by Ahmadi et al. [62]. Fe0 can react with O2 under acidic condition to form H2O2 (Eq. (31)). Then, Fe2+ activate the H2O2 to generate the HO· in the solution (Eq. (32)), improving the free radical concentration. Jaafarzadeh et al. [84] compared the decolorization of acid brown 14 by photoelectro-oxidation/PMS and photo-electro-oxidation/PMS/transitional metals ion (Mn+) (M = Cu2+, Fe2+ and Co2+), respectively. The presence of transitional metals ions increased the decolorization of AB14 for the more generation of SO4·- and HO· (Eqs. (33) and (34)). Wang and his colleague [56] adopted iron plate as a sacrificial anode and studied the degradation process of 2, 4, 5-trichlorophenol, which shows that Fe2+ would release from the anode and get into the reaction solution. And then, the involvement of UV irradiation enhanced the regeneration of Fe2+ (Eq. (35)), accelerating the generation of SO4·- and HO·.
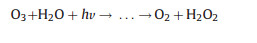
|
(30) |

|
(31) |

|
(32) |

|
(33) |
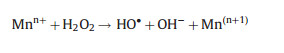
|
(34) |

|
(35) |
The comparative study of the different (photo)EAOP-oxidants systems was shown in Table 4, which shown that the EAOP-oxidants have an effective degradation efficiency for the various organics with the different technical features. The addition of oxidants (e.g., PS, PMS, and O3) in the electrochemical advanced oxidation process highly improved the generated concentration of radicals (e.g., sulfate radical and hydroxyl radical). In general, the EAOPs-PS/PMS/O3 system generates free radicals mainly through the electron transfer of PS/PMS on cathode and PS/ PMS activation based on the transition metal ion or metal oxides. Furthermore, when the cathode material was carbon-based materials, the hydrogen peroxide could generate through the cathode reduction of O2 and be activated by the activators to produce hydroxyl radical. Based on the EAOPs-PS/PMS/O3 system, the presence of UV light further strengthens the reaction process through accelerating oxidants activation to generate radicals and facilitating the Fe2+ regeneration from the iron complexes. It is the synergistic effect among electrolysis, oxidants, catalyst and/or light promotes the degradation of pollutants. However, it is worth mentioning that the optimal parameters conditions in the EAOPs-oxidants coupling systems were vital to prevent the radical self-consuming reactions.
|
|
Table 4 The comparative study of the different (photo)EAOP-oxidants. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
7. Conclusions
In summary, the review provided effective methods based on the current development of coupling technology about the electrochemical advanced oxidation process and the oxidants activation for the organic compound degradation. As stated in this review, we summarize the electro-PS activation, the homogeneous EAOPs-PS system with Fe2+/Fe3+ as the catalyst, and the heteroge-neous EAOPs-PS system with the single-metal oxide or mixed-metal oxides as activators, and make a comprehensive analysis of the reaction mechanism. It is worth noting that the reaction mechanisms of EAOPs-PMS coupling systems are similar to the EAOPs-PS system and the efficient degradation of pollutants is due to the synergistic effect of the individual process. In addition, the EAOPs-peroxone systems used for various pollutants were intro-duced and we make a comparison of the source of H2O2 with the diverse cathode materials. Also, the different reaction mechanisms of EAOPs-peroxone system and electrolysis-ozone system were concluded, respectively. The degradation process and the effect of electrolyte on pollutant removal were discussed in detail. Finally, the photoelectro-oxidant systems were introduced comprehen-sively. In conclusion, (photo)EAOPs-oxidants as environment-friendly coupling systems can fully exert synergistic catalysis during the reaction process, significantly improving the degrada-tion and mineralization efficiency.
AcknowledgmentsThe authors would like to acknowledge the financial support from the National Natural Science Foundation of China (No. 51878423) and Graduate Student's Research and Innovation Fund of Sichuan University (No. 2018YJSY075).
| [1] |
F.C. Moreira, R.A.R. Boaventura, E. Brillas, V.J.P. Vilar, Appl. Catal. B:Environ. 202 (2017) 217-261. DOI:10.1016/j.apcatb.2016.08.037 |
| [2] |
M. Iram, C. Guo, Y. Guan, A. Ishfaq, H. Liu, J. Hazard. Mater. 181 (2010) 1039-1050. DOI:10.1016/j.jhazmat.2010.05.119 |
| [3] |
Y. Gan, X. Wang, L. Zhang, et al., Chemosphere 218 (2019) 860-868. DOI:10.1016/j.chemosphere.2018.11.192 |
| [4] |
B. Lai, Y. Zhou, J. Wang, Y. Zhang, Z. Chen, Environ. Technol. 35 (2014) 973-983. DOI:10.1080/09593330.2013.857700 |
| [5] |
B. Lai, Y.X. Zhou, P. Yang, J.H. Yang, J.L. Wang, Chemosphere 90 (2013) 1470-1477. DOI:10.1016/j.chemosphere.2012.09.040 |
| [6] |
B. Lai, Y.X. Zhou, P. Yang, Chem. Eng. J. 200- 202 (2012) 10-17. |
| [7] |
B.C. Hodges, E.L. Cates, J.H. Kim, Nat. Nanotechnol. 13 (2018) 642-650. DOI:10.1038/s41565-018-0216-x |
| [8] |
X. Yang, X. Cheng, A.A. Elzatahry, et al., Chin. Chem. Lett. 30 (2019) 324-330. DOI:10.1016/j.cclet.2018.06.026 |
| [9] |
K. Chen, G.H. Wang, W.B. Li, et al., Chin. Chem. Lett. 25 (2014) 1455-1460. DOI:10.1016/j.cclet.2014.06.014 |
| [10] |
Q. Ji, J. Li, Z. Xiong, B. Lai, Chemosphere 172 (2017) 10-20. DOI:10.1016/j.chemosphere.2016.12.128 |
| [11] |
Z. Xiong, B. Lai, P. Yang, et al., J. Hazard. Mater. 297 (2015) 261-268. DOI:10.1016/j.jhazmat.2015.05.006 |
| [12] |
L. Lai, J. Yan, J. Li, B. Lai, Chem. Eng. J. 343 (2018) 676-688. DOI:10.1016/j.cej.2018.01.035 |
| [13] |
Y.Q. Zhang, W.L. Huang, D.E. Fennell, Chin. Chem. Lett. 21 (2010) 911-913. DOI:10.1016/j.cclet.2010.03.001 |
| [14] |
Z. Xiong, B. Lai, Y. Yuan, et al., Chem. Eng. J. 302 (2016) 137-145. DOI:10.1016/j.cej.2016.05.052 |
| [15] |
R. Munter, Proc. Estonian Acad. Sci. Chem. 50 (2001) 59-80. |
| [16] |
R. Andreozzi, V. Caprio, A. Insola, R. Marotta, Catal. Today 53 (1999) 51-59. DOI:10.1016/S0920-5861(99)00102-9 |
| [17] |
J. Li, Q. Liu, Ji Qq., B. Lai, Appl. Catal. B:Environ. 200 (2017) 633-646. DOI:10.1016/j.apcatb.2016.07.026 |
| [18] |
H. Zhang, Q. Ji, L. Lai, G. Yao, B. Lai, Chin. Chem. Lett. 30 (2019) 1129-1132. DOI:10.1016/j.cclet.2019.01.025 |
| [19] |
J. Li, Y. Ren, F. Ji, B. Lai, Chem. Eng. J. 324 (2017) 63-73. DOI:10.1016/j.cej.2017.04.104 |
| [20] |
Q. Zhao, Q. Mao, Y. Zhou, et al., Chemosphere 189 (2017) 224-238. DOI:10.1016/j.chemosphere.2017.09.042 |
| [21] |
Y. Qin, M. Sun, H. Liu, J. Qu, Electrochim Acta 186 (2015) 328-336. DOI:10.1016/j.electacta.2015.10.122 |
| [22] |
Y.J. Feng, X.Y. Li, Water Res. 37 (2003) 2399-2407. DOI:10.1016/S0043-1354(03)00026-5 |
| [23] |
S. Garcia-Segura, J.D. Ocon, M.N. Chong, Process. Saf. Environ. 113 (2018) 48-67. DOI:10.1016/j.psep.2017.09.014 |
| [24] |
W.S. Chen, Y.C. Jhou, C.P. Huang, Chem. Eng. J. 252 (2014) 166-172. DOI:10.1016/j.cej.2014.05.033 |
| [25] |
Z. Liu, C. Zhao, P. Wang, et al., Chem. Eng. J. 343 (2018) 28-36. DOI:10.1016/j.cej.2018.02.114 |
| [26] |
W.S. Chen, C.P. Huang, Chemosphere 125 (2015) 175-181. DOI:10.1016/j.chemosphere.2014.12.053 |
| [27] |
L. Zhang, W. Ding, J. Qiu, et al., J. Clean. Prod. 197 (2018) 297-305. DOI:10.1016/j.jclepro.2018.05.267 |
| [28] |
S. Wacawek, V. Antos, P. Hrabak, M. Cernik, D. Elliott, Environ. Sci. Pollut. R. 23 (2016) 765-773. DOI:10.1007/s11356-015-5312-y |
| [29] |
J.E. Silveira, A.L. Garcia-Costa, T.O. Cardoso, J.A. Zazo, J.A. Casas, Electrochim Acta 258 (2017) 927-932. DOI:10.1016/j.electacta.2017.11.143 |
| [30] |
J. Liu, S. Zhong, Y. Song, B. Wang, F. Zhang, J. Electroanal. Chem. 809 (2018) 74-79. DOI:10.1016/j.jelechem.2017.12.033 |
| [31] |
W.S. Chen, C.P. Huang, Chem. Eng. J. 266 (2015) 279-288. DOI:10.1016/j.cej.2014.12.100 |
| [32] |
H. Zhang, Z. Wang, C. Liu, et al., Chem. Eng. J. 250 (2014) 76-82. DOI:10.1016/j.cej.2014.03.114 |
| [33] |
J. Wu, H. Zhang, J. Qiu, J. Hazard. Mater. 215 (2012) 138-145. |
| [34] |
J.E. Silveira, T.O. Cardoso, M. Barreto-Rodrigues, J.A. Zazo, J.A. Casas, Environ. Technol. 39 (2018) 1208-1216. DOI:10.1080/09593330.2017.1323960 |
| [35] |
M.R. Samarghandi, J. Mehralipour, G. Azarian, K. Godini, A. Shabanlo, Global Nest. J. 19 (2017) 115-121. DOI:10.30955/gnj.002063 |
| [36] |
Y. Long, Y. Feng, X. Li, et al., Chemosphere 219 (2019) 1024-1031. DOI:10.1016/j.chemosphere.2018.12.054 |
| [37] |
A. Long, H. Zhang, Environ. Sci. Pollut. R. 22 (2015) 11606-11616. DOI:10.1007/s11356-015-4406-x |
| [38] |
J. Li, Y. Ren, L. Lai, B. Lai, J. Hazard. Mater. 344 (2018) 778-787. DOI:10.1016/j.jhazmat.2017.11.007 |
| [39] |
X. Huang, D. An, J. Song, W. Gao, Y. Shen, J. Clean. Prod. 165 (2017) 637-644. DOI:10.1016/j.jclepro.2017.07.171 |
| [40] |
A. Ledjeri, I. Yahiaoui, F. Aissani-Benissad, J. Environ. Manage. 184 (2016) 249-254. DOI:10.1016/j.jenvman.2016.09.086 |
| [41] |
A. Ledjeri, I. Yahiaoui, H. Kadji, et al., Environ. Toxicol. Phar. 53 (2017) 34-39. DOI:10.1016/j.etap.2017.04.022 |
| [42] |
H. Lin, J. Wu, H. Zhang, Sep. Purif. Technol. 117 (2013) 18-23. DOI:10.1016/j.seppur.2013.04.026 |
| [43] |
C. Yang, B. Ren, D. Wang, Q. Tang, Environ. Technol. (2019) 1-14. |
| [44] |
S. Yan, W. Xiong, S. Xing, et al., Sci. Total. Environ. 599 (2017) 1181-1190. |
| [45] |
Y. Xu, H. Lin, Y. Li, H. Zhang, Sci. Total. Environ. 609 (2017) 644-654. DOI:10.1016/j.scitotenv.2017.07.151 |
| [46] |
H. Lin, X. Zhong, C. Ciotonea, et al., Appl. Catal. B-Environ. 230 (2018) 1-10. DOI:10.1016/j.apcatb.2018.02.014 |
| [47] |
H. Lin, H. Zhang, L. Hou, J. Hazard. Mater. 276 (2014) 182-191. DOI:10.1016/j.jhazmat.2014.05.021 |
| [48] |
H. Lin, Y. Li, X. Mao, H. Zhang, J. Taiwan. Inst. Chem. Eng. 65 (2016) 390-398. DOI:10.1016/j.jtice.2016.05.050 |
| [49] |
B. Deng, Y. Li, W. Tan, et al., Chemosphere 204 (2018) 178-185. DOI:10.1016/j.chemosphere.2018.03.194 |
| [50] |
C. Cai, Z. Zhang, H. Zhang, J. Hazard. Mater. 313 (2016) 209-218. DOI:10.1016/j.jhazmat.2016.04.007 |
| [51] |
C. Cai, H. Zhang, X. Zhong, L. Hou, Water Res. 66 (2014) 473-485. DOI:10.1016/j.watres.2014.08.039 |
| [52] |
F. Sepyani, R.D.C. Soltani, S. Jorfi, H. Godini, M. Safari, J. Environ. Manage. 224 (2018) 315-326. DOI:10.1016/j.jenvman.2018.07.072 |
| [53] |
J. Li, J. Yan, G. Yao, et al., Chem. Eng. J. 361 (2019) 1317-1332. DOI:10.1016/j.cej.2018.12.144 |
| [54] |
H. Lin, J. Wu, H. Zhang, Chem. Eng. J. 244 (2014) 514-521. DOI:10.1016/j.cej.2014.01.099 |
| [55] |
N. Jaafarzadeh, F. Ghanbari, M. Alvandi, Sustainable Environ. Res. 27 (2017) 223-229. DOI:10.1016/j.serj.2017.06.001 |
| [56] |
Y.R. Wang, W. Chu, Chem. Eng. J. 215 (2013) 643-650. |
| [57] |
Y.R. Wang, W. Chu, Water Res. 45 (2011) 3883-3889. DOI:10.1016/j.watres.2011.04.034 |
| [58] |
S. Yan, J. Geng, R. Guo, Y. Du, H. Zhang, J. Hazard. Mater. 333 (2017) 358-368. DOI:10.1016/j.jhazmat.2017.03.043 |
| [59] |
J.P. Zou, Y. Chen, S.S. Liu, et al., Water Res. 150 (2019) 330-339. DOI:10.1016/j.watres.2018.11.077 |
| [60] |
J. Li, H. Lin, K. Zhu, H. Zhang, Chemosphere 188 (2017) 139-147. DOI:10.1016/j.chemosphere.2017.08.137 |
| [61] |
M. Arellano, Angeles Sanroman M., M. Pazos, Sep. Purif. Technol. 208 (2019) 34-41. DOI:10.1016/j.seppur.2018.05.028 |
| [62] |
M. Ahmadi, F. Ghanbari, J. Environ. Manage. 228 (2018) 32-39. DOI:10.1016/j.jenvman.2018.08.102 |
| [63] |
B. Bakheet, S. Yuan, Z. Li, et al., Water Res. 47 (2013) 6234-6243. DOI:10.1016/j.watres.2013.07.042 |
| [64] |
W. Guo, Q.L. Wu, X.J. Zhou, et al., RSC Adv. 5 (2015) 52695-52702. DOI:10.1039/C5RA07951A |
| [65] |
X. Li, G. Liu, M. Shi, et al., Sep. Purif. Technol. 165 (2016) 154-159. DOI:10.1016/j.seppur.2016.03.048 |
| [66] |
X. Li, Y. Wang, S. Yuan, et al., Water Res. 63 (2014) 81-93. DOI:10.1016/j.watres.2014.06.009 |
| [67] |
H. Wang, S. Yuan, J. Zhan, et al., Water Res. 80 (2015) 20-29. DOI:10.1016/j.watres.2015.05.024 |
| [68] |
M. Hou, Y. Chu, X. Li, et al., J. Hazard. Mater. 319 (2016) 61-68. DOI:10.1016/j.jhazmat.2015.12.054 |
| [69] |
X. Li, Y. Wang, J. Zhao, et al., J. Hazard. Mater. 300 (2015) 298-306. DOI:10.1016/j.jhazmat.2015.07.004 |
| [70] |
C. Qu, S. Lu, D. Liang, et al., J. Hazard. Mater. 364 (2019) 468-474. DOI:10.1016/j.jhazmat.2018.10.073 |
| [71] |
A. Fischbacher, J. von Sonntag, C. von Sonntag, T.C. Schmidt, Environ. Sci. Technol. 47 (2013) 9959-9964. DOI:10.1021/es402305r |
| [72] |
Y. Zhang, S. Zuo, Y. Zhang, et al., J. Hazard. Mater. 368 (2019) 771-777. DOI:10.1016/j.jhazmat.2019.02.005 |
| [73] |
Y. Wang, G. Yu, S. Deng, J. Huang, B. Wang, Chemosphere 208 (2018) 640-654. DOI:10.1016/j.chemosphere.2018.05.095 |
| [74] |
G. Xia, Y. Wang, B. Wang, et al., Water Res. 118 (2017) 26-38. DOI:10.1016/j.watres.2017.04.005 |
| [75] |
Z. Guo, Y. Xie, Y. Wang, et al., Chem. Eng. J. 337 (2018) 733-740. DOI:10.1016/j.cej.2017.11.178 |
| [76] |
O. Turkay, S. Barisci, B. Ozturk, et al., J. Electrochem. Soc. 164 (2017) E180-E186. DOI:10.1149/2.0431709jes |
| [77] |
C. Rosales Landeros, C.E. Barrera Diaz, A. Amaya Chaves, G. Roa Morales, Fuel 198 (2017) 91-98. DOI:10.1016/j.fuel.2016.10.031 |
| [78] |
Z. Xiong, B. Lai, P. Yang, Water Res. 140 (2018) 12-23. DOI:10.1016/j.watres.2018.04.030 |
| [79] |
H. Wang, B. Bakheet, S. Yuan, et al., J. Hazard. Mater. 294 (2015) 90-98. DOI:10.1016/j.jhazmat.2015.03.058 |
| [80] |
S.S. Liu, Q.J. Xing, Y. Chen, et al., ACS Sustain. Chem. Eng. 7 (2019) 1250-1259. DOI:10.1021/acssuschemeng.8b04917 |
| [81] |
N. Bensalah, A. Bedoui, Environ. Technol. 38 (2017) 2979-2987. DOI:10.1080/09593330.2017.1284271 |
| [82] |
N. Jaafarzadeh, G. Barzegar, F. Ghanbari, Process. Saf. Environ. 111 (2017) 520-528. DOI:10.1016/j.psep.2017.08.012 |
| [83] |
W. Shen, Y. Wang, J. Zhan, et al., Chem. Eng. J. 310 (2017) 249-258. DOI:10.1016/j.cej.2016.10.111 |
| [84] |
N. Jaafarzadeh, F. Ghanbari, M. Moradi, Korean J. Chem. Eng. 32 (2015) 458-464. DOI:10.1007/s11814-014-0263-4 |
| [85] |
Z. Lin, W. Yao, Y. Wang, et al., Water Res. 88 (2016) 691-702. DOI:10.1016/j.watres.2015.11.005 |