 2019, Vol. 30
2019, Vol. 30 


b School of Chemistry and Chemical Engineering, University of South China, Hengyang 421001, China;
c Department of Chemistry, Hunan University of Science and Technology, Xiangtan 411201, China
The concept of "Green Chemistry" was raised in 1990s due to the increasing attention to chemical pollution and resource depletion. It is defined as the "design of chemical products and processes to reduce or eliminate the use and generation of hazardous substances"In 1998, Anastas and Warner published the Twelve Principles of Green Chemistry, which then was summarized as the convenient acronym "productivity" in 2005. Nowadays, the principles of green chemistry have been widely adopted as guidelines in the design of green and sustainable chemical process. For example, the utilization of green solvents (such as water [1], biomass solvent [2], ionic liquids [3], and deep eutectic solvents [4]), nontoxic and inexpensive catalysis (e.g., organocatalysis, biocatalysis), green chemical technologies (e.g., visible-light photochemistry [5], sonochemistry [3b, 6]) to minimize the production of wastes from organic reactions have emerged as commonly used strategies for green chemistry research.
During the past few years, our group has devoted ourselves to the development of green and sustainable synthetic methodolo-gies for the construction of valuable compounds from easily available starting materials based on the design of multifunctional reactants/reagents or reaction media, through the assistance of green chemistry technology such as ultrasonic irradiation. Herein, we summarized some of our observation in green and sustainable chemistry, which demonstrated that the concept of dual roles design (i.e., two birds one stone strategy) is useful for the development of for green organic preparation. In those design, the reactant, catalyst or solvent may play dual roles rendering a cleaner organic synthesis process.
2. Dual roles of solvents 2.1. Water as the reaction media and reactantWater is a natural abundant, nontoxic, low-cost, easily available green solvent. More importantly, water could not only serve as a green reaction media but also display unique properties to promote the organic transformations. Sulfur-containing com-pounds play a vital role in fine chemicals, medicinal and materials chemistry [7]. Among them, the β-sulfonyl-α, β-unsaturated carbonyl compounds are important sulfur-containing compounds present in a wide range of natural products and synthetic drugs. Most of the previous reported procedures for the construction of these compounds were conducted in organic solvents. Among them, E-isomers were found to be the major products. The selective synthesis of Z-isomers remains a challenging task, which is less thermodynamically favorable. In 2016, we reported an efficient and simple sulfonylation reaction [8] of activated alkynes 1a with sodium sulfinates 1b in water without any additives (Scheme 1) [9]. This procedure showed broad substrate scope and high Z-stereoselectivity. The investigation of mechanism revealed that the sulfonylation reaction proceeded through the water promoted anti-addition of alkynylcarbonyl compounds [10] furnishing high Z-selectivity. In this protocol, water was applied as green solvent and also hydrogen source, which was confirmed by the isotope labeling experiment in D2O.

|
Download:
|
| Scheme 1. Selective preparation of Z-β-sulfonyl enoates. | |
{kind=link}
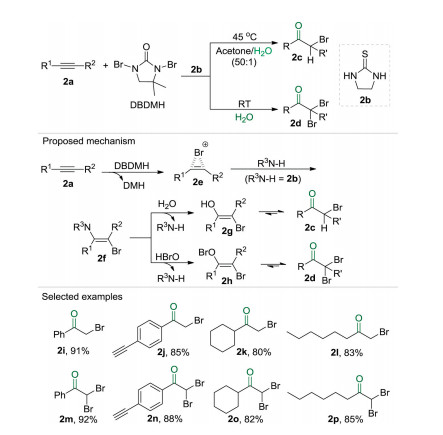
α-Halo and α, α'-dihalo ketones are valuable intermediates for organic synthesis [11]. Despite significant progress has been achieved, the synthesis of haloketones, the direct synthesis of α-halo and α, α'-dihalo ketones from unactivated alkyne and green halogenating reagents under metal-free, additive-free and oxi-dant-free conditions is an attractive procedure with high atom economy and step economy. Fortunately, by using the cheap and readily available 1, 3-dibromo-5, 5-dimethyldantoin (DBDMH) as halogen source and thiourea 2b as catalyst, a highly selective and controllable catalytic system was established for the three-component reaction of unactivated alkynes 2a, DBDMH and water to synthesize α-halo and α, α'-dihalo ketones (2c and 2d) (Scheme 2) [12]. In this catalytic system, the selectivity of the reaction could be tuned by simply changing the reaction solvent (mixture of acetone/ water or water) and temperature (45 ℃ or room temperature) delivering products 2c or 2d respectively. Furthermore, the isotope labeling experiment confirmed that H218O was the oxygen source in these transformations.

|
Download:
|
| Scheme 2. Selective preparation of α-mono or α, α'-dihalo ketones. | |
{kind=link}
Based on the experimental observations, a plausible mechanism was proposed. Firstly, the brominating of alkyne moiety 2a by DBDMH gave bromirenium 2e along with 5, 5-dimethylhydantoin (DMH). Then an anti-addition of the amino group of thiourea to intermediate 2e delivered the β-haloen-amine 2f as a key intermediate. When the reaction was conducted in the mixture of acetone and water, the substitution of the amino group on 2f with the hydroxyl ion of water afforded the enol 2g, followed by a tautomerization producing the product α-halo ketone 2c. Alternatively, when water was applied as the sole solvent, the in situ generated HOBr might act as a nucleophile to react with β-haloenamine 2f affording intermediate 2h, which was converted into α, α'-dihalo ketone 2d via a tautomerization. Notably, in this protocol, a dual roles concept was verified, e.g., the bromine source DBDMH also serves as an activator for alkynes, and the solvent water could simultaneously function as an oxygen source.
2.2. Eco-friendly ether as the reaction media and promoterThe selective oxidation of alcohols into the corresponding ketones or carboxylic acids is an important transformation in organic synthesis. In such a selective oxidation reaction, a stoichiometric amount of oxidant is generally necessary. Obvious-ly, molecular oxygen is one of the cleanest and easily available oxidants [13]. However, the application of atmospheric pressure of O2 is challenging due to its inherent inertness. In 2018, our group developed a simple metal- and base-free system for the selective oxidation of aromatic alcohols 3a into the corresponding aromatic carboxylic acids 3c or aromatic ketones 3d using the high boil point bis(methoxypropyl) ether 3b as reaction media in the presence of atmospheric pressure of O2 under 120 ℃ (Scheme 3) [14]. Notably, this oxidative system has displayed excellent functional-group tolerance and scalable potential (up to 50 g-scale) under mild conditions. Moreover, various experiments including the kinetic isotope effect (KIE) experiment, electron paramagnetic resonance (EPR) experiment, and control experiments using isotopic labelled 18O2 have demonstrated that the cleavage of the benzylic C(sp3)-H bond is the rate-determining step and the cleavage of C(sp3)-O bond of alcohol should be involved. Based on these observations, a plausible mechanism was proposed. Firstly, the ether 3b reacted with O2 to produce a hydroperoxide, which further oxidized the substrate 3a affording the corresponding radical 3e and a hydroxyl radical. Then the coupling of 3e and hydroxyl radical generated a diol intermediate 3f, which was converted into the carbonyl compound 3d (aromatic aldehyde or ketone) and water. In the case of aldehyde 3d (R =H), a radical autoxidation is possible to generate the aromatic carboxylic acid 3c as the final product. In this reaction, the bis(methoxypropyl) ether 3b was the reaction media and also a radical initiator in the presence of O2.

|
Download:
|
| Scheme 3. Selective oxidation of alcohols. | |
{kind=link}
2.3. Ionic liquid as the reaction media and promoter
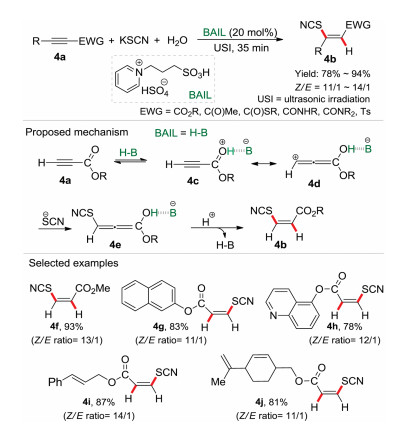
In our previous work [9], it was found that water was the efficient hydrogen source for the hydrosulfonylation of activated alkynes (Scheme 1). However, hydrothiocyanation of activated alkynes with water as the hydrogen source was unexplored under eco-friendly reaction conditions. On the other hand, ionic liquids have been recognized as a class of green solvent and versatile task-specific reagents for organic synthesis in the past decades [15]. In 2018, a Brønsted acidic ionic liquid (BAIL) catalytic system was established for the hydrothiocyanation of activated alkynes 4a with KSCN and water under the irradiation of ultrasound (Scheme 4) [3b]. Comparing with the conventional heating-conditions, the ultrasonic radiation could enhance the reaction rate and selectivity furnishing the desired product with high yield (78%–94%) in 35 min. In this procedure, various Z-vinyl thiocya-nates were synthesized in good to excellent yields, and the BAIL catalyst could be recycled and reused at least for 5 times without significant loss of activities. For the reaction mechanism, it is proposed that the terminal alkyne 4a was firstly protonated by the BAIL, which then generated the zwitterionic intermediate 4d via tautomerization. Then the nucleophilic thiocyanate ion (SCN-) attacked the β-carbon of intermediate 4d to produce the intermediate 4e, followed by protonation affording the Z-vinyl thiocyanate 4b. Remarkably, in this procedure the Brønsted acidic ionic liquid was an efficient promoter and also a co-solvent for the hydrothiocyanation reaction.

|
Download:
|
| Scheme 4. Hydrothiocyanation of activated alkynes promoted by ionic liquid. | |
{kind=link}
2.4. Natural deep eutectic solvents as the reaction media and promoter
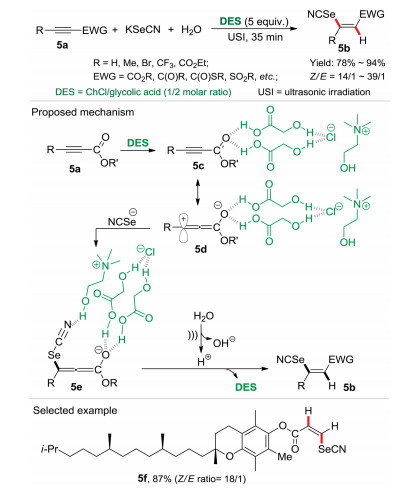
Natural deep eutectic solvents (DESs) are bio-degradable, non-cytotoxic and easy preparation, which made from entirely of renewable plant metabolites. During our continuous investigation in sustainable chemistry, we found that natural DESs in organic synthesis not only served as green solvents but also significant promoters for the organic transformations. For example, in the natural deep eutectic mixture of choline chloride (ChCl) and glycolic acid (ChCl/glycolic acid, mol ratio = 1:2), the one-pot procedure for the construction of Z-vinyl selenolates 5b was successfully established under the ultrasonic irradiation (USI, 35 W/40 kHz) for 35 min via a three-components reaction of activated alkynes 5a, KSeCN, and water (Scheme 5) [4c]. In this selenocyanation reaction, the ultrasonic irradiation is significant for the high reactivity. It could be assumed that the active cavitation bubbles generated during the ultrasonic irradiation will be advantageous to the mass transfer process, resulting in an enhanced performance of the selenocyanation reaction. The mechanism studies demonstrated that the hydrogen bonds between the solvent DES and the substrate alkyne 5a may enhance the polarization of carbonyl group (5c), resulting a resonance structure in zwitterionic form, i.e. intermediate 5d. Subsequently, 5d underwent the addition of selenocyanate anion giving the intermediate 5e, in which the hydrogen bond between "SeCN" and hydroxyl group of ChCl in DES was formed. On the other hand, protons could be generated by self-ionization of H2O with the aid of ultrasound irradiation. Finally, the protonation of intermediate 5e on the less sterically hindered side delivered the desired product Z-vinyl selenocyanates 5b. Notably, this one-pot reaction strategy showed broad substrate scope with a wide range of alkynes, and it also worked well for large-scale synthesis. In this protocol, DES as a green reaction media and promoter via hydrogen bonding, could be recycled and reused at least for 5 times without significant loss of activities.

|
Download:
|
| Scheme 5. Selenocyanation reaction of alkynes in DES. | |
{kind=link}
3. Dual roles of reactant 3.1. Reaction intermediate as the reactant and activator
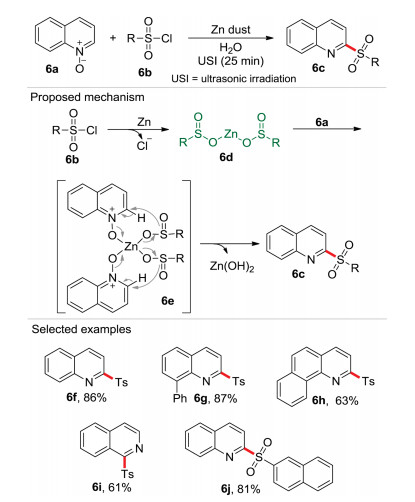
C2-functionalized quinoline is a commonly found nitrogen heterocycle [16] in biologically active natural products and unnatural pharmaceuticals [17]. For this reason, it is not surprising that since Cui-Wu coupling reaction pioneered in 2009 [18], numerous protocols have been reported for the construction and functionalization of such skeleton [19]. For the construction of 2-sulfonylquinolines from quinoline N-oxides and sulfonyl chloride, the only two previous methods were reported employing the organic solvents and toxic reagents [20]. In 2017, our group reported an ultrasound-assisted one-pot C2-sulfonylation reaction of quinoline N-oxide 6a with sulfonyl chloride 6b promoted by Zn powder using water as solvent (Scheme 6) [6]. In this process, the sulfonyl chloride 6b was firstly reduced by Zn dust to generate a zinc bissulphinate intermediate 6d, which then coordinated with quinoline N-oxide 6a affording the intermediate 6e. Then 6e was converted into the target compound 6c and zinc hydroxide via an intramolecular nucleophilic addition. Notably, the sulfur charge reversion (6b vs. 6e) was observed in this procedure. In this reaction, an atom economy of 70.7%, an E-factor of 1.17 and an eco-scale score of 71 were obtained, revealing that this process is remarkably greener than the previous routes. The in situ generated zinc bissulphinate intermediate 6d was reactant and activator for the quinoline N-oxide 6a.

|
Download:
|
| Scheme 6. One-pot C2-sulfonylation of quinoline N-oxides. | |
{kind=link}
3.2. Reactant as the reaction partner and activator
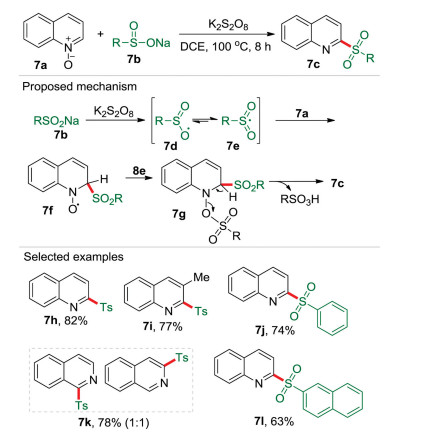
As mentioned above, quinolines are important heterocyclic scaffolds which are ubiquitous in various biologically active compounds and pharmaceuticals. Therefore, the synthesis of quinolines derivatives from easily available starting materials has drawn huge attention. Quinoline N-oxide is one of the most useful substrates for the development of synthetic methodologies to construct valuable quinolines derivatives. Consequently, the activation of quinoline N-oxides for functionalization of quinolines has been well developed. However, most of the previous works were conducted through nucleophilic addition pathway. The only reported radical process for the deoxygenative 2-sulfonylation of quinoline N-oxides required a copper catalyst under an argon atmosphere [21]. In 2018, we disclosed a transition-metal-free radical coupling reaction of quinoline N-oxides 7a and sodium sulfinates 7b to afford a range of 2-sulfonylquinoline derivatives 7c by using K2S2O8 as the sole promoter in 1, 2-dichloroethane (DCE) (Scheme 7) [22]. Based on the experimental investigation including EPR, a radical mechanism was proposed. It is believed that single electron oxidation of sodium sulfinate 7b produce the oxygen-centered radical 7d, which resonated with the sulfonyl radical 7e. Subsequently, an intermediate 7f could be generated via a Minisci-like radical addition of radical 7e to quinoline N-oxide 7a. The intermediate 7f further coupled with the sulfur-centered sulfate radical 7e to generate the intermediate 7g, which then was converted into 2-sulfonylquinoline 7c due to the aromatization driving force with releasing sulfonic acid. In this reaction, sodium sulfinate plays dual roles of an oxidant and an activating agent (in 7g).

|
Download:
|
| Scheme 7. Deoxygenative 2-sulfonylation of quinoline N-oxides. | |
{kind=link}
For the deoxygenative 2-amidation of quinoline N-oxide, the previous reported activators are generally electrophilic reagents, e.g., H-phosphonate, iodine reagents, boron reagents, etc. However, radical type activator is less explored. As part of our ongoing interest in green chemistry, we recently reported a radical activation approach to realize the deoxygenative 2-amidation of quinoline N-oxides 8a with nitriles 8b to synthesize N-acylated 2-aminoquinolines 8c, in which Selectfluor was applied as an oxidant (Scheme 8) [23]. It was proposed that methyl carbazate (NH2NHCO2Me) was firstly oxidized by Selectfluor via a single electron transfer (SET) process affording the radical 8d. The addition of radical 8d to quinoline N-oxide gave a radical cation 8e, followed by a single electron oxidation to produce quinolinium intermediate 8f. Subsequently, the α-carbon of intermediate 8f suffer from a nucleophilic addition of nitrile 8b resulting the intermediate 8g, which underwent deprotonation to form the dehydro-aromatization intermediate 8h. Finally, the decomposi-tion of 8hgenerated the benzimidic acid 8i, followed by a rapidly tautomerization to produce the more stable 8c. By using this novel system, a series of substrates bearing valuable functional groups could be converted into the corresponding N-acylated 2-amino-quinoline derivatives in good to excellent yields. Notably, in this metal-, base- and reductant-free procedure the ester methyl carbazate (NH2NHCO2Me) was demonstrated to be a radical precursor and also an oxygen source.

|
Download:
|
| Scheme 8. Deoxygenative 2-amidation of quinoline N-oxides. | |
{kind=link}
We reported a direct synthesis of diverse heteroaromatic derivatives through Selectfluor-mediated regioselective nucleo-philic functionalization of N-heterocycles [16a, 16o, 24] 9a under metal- and base-free and open-air conditions (Scheme 9) [25]. In this protocol, the radical cation 9d was formed via a single-electron-transfer process between quinoline 9a and Selectfluor along with the formation of fluorine radical. Then, the nucleophile attacked the C-2 position of radical cation 9d to afford the intermediate 9e, which subsequently underwent radical-coupling reaction with fluorine radical to yield the intermediate 9f (path a). Alternatively, quinoline 9a reacted with Selectfluor to produce the active N-fluoroquinolinium intermediate 9g, which subsequently underwent nucleophilic addition of a nucleophile to give intermediate 9f (path b). Finally, elimination of HF from intermediate 9f regenerated the aromatic ring to deliver the final product 9c. Through this facile, practical and environment-friendly method, a broad range of nucleophilic functionalization of N-heterocycles 9c were prepared in moderated to excellent yields. In addition, the gram-scale experiment was carried out and the desired product was obtained in good yield, demonstrating the promising application in industry. In this system, Selectfluor was an activator for the substrates 9a and the in situ generated fluorine radical also played an important role to form the intermediate 9f.

|
Download:
|
| Scheme 9. Selectfluor-mediated functionalization of N-heterocycles. | |
{kind=link}
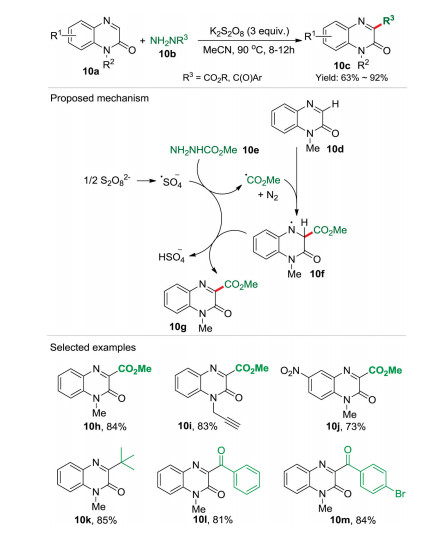
During the past few years, remarkable progress has been achieved on the direct functionalization of quinoxalin-2(1H)-ones, As part of our sustained efforts in the development of ecofriendly synthetic methodology in organic synthesis, the green introduc-tion of an ester group into quinoxalin-2(1H)-ones was developed recently (Scheme 10) [26]. By employing carbazates 10b as the eco-friendly ester source, 30 examples of C3-alkoxycarbonylation of quinoxalin-2(1H)-ones were achieved in moderated to excellent yields. More importantly, multiple functional groups, especially alkynyl, can be tolerated due to the mild reaction conditions. In the proposed mechanism, the hemolytic-bond-cleavage of persulfate generated sulfate radical anion, which reacted with the methyl carbazate 10e to form the ester free-radical along with HSO4 anion and molecular nitrogen. Afterward, the addition of ester free-radical to reaction substrate 10d lead to the formation of free-radical intermediate 10f, which further underwent hydrogen abstraction/oxidation to regenerate the aromatic ring furnishing the final product 10g. Remarkably, this synthetic methodology was featured innocuous nitrogen as the byproduct, wide reaction substrates, metal- and base-free conditions as well easy scale-up ability. It should be noted that the reactant carbazate was an ester source, and also a radical precursor to deliver radicals triggering the target reaction.

|
Download:
|
| Scheme 10. 3-Alkylatoin/acylation of quinoline N-oxides. | |
{kind=link}
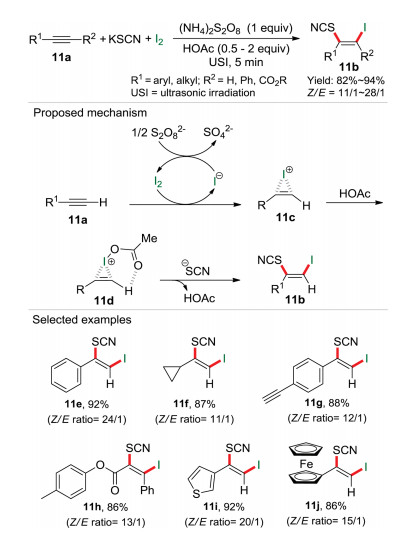
In 2019, we developed a metal- and solvent-free ultrasonic method for synthesis of diverse (Z)-β-iodo vinylthiocyanates through multicomponent reaction [27] of alkynes, KSCN and molecular iodine (Scheme 11) [28]. As it can be seen in the proposed reaction mechanism, alkyne 11a react with I2 to produce activated iodoirenium ion 11c along with iodine anion, which was further oxidized to I2 by (NH4)2S2O8. Next, an energetically favorable six-membered ring 11d was proposed due to the hydrogen bond between HOAc and intermediate 11c. Finally, 11d was attacked by the thiocyanate anion at the less steric hindered carbon atom to yield the desired product 11b. Notably, diverse alkynes, including wide range of arylacetylenes as well as aliphatic acetylenes with different chain lengths and various heteroatomic functional groups, were all react smoothly to give the desired products in moderate to excellent yields. Moreover, when 1, 4-diethynylbenzene, which contains two alkynyl groups, was employed as reaction substrate, only monoiodothiocyanation product 11g was obtained and no double addition product could be detected. Additionally, ferrocenylacetylene was also suitable for this elegant transformation, which highlighted the importance of mild reaction conditions of this metal-free and solvent-free ultrasonic system. Similarly, in this procedure iodine is not only a reactant but also an important activator for the alkynes.

|
Download:
|
| Scheme 11. Metal- and solvent-free synthesis of Z-β-iodo vinylthiocyanates. | |
{kind=link}
4. Conclusions
Owning to the advantages of green and sustainable chemistry, enormous efforts have been devoted to the development of green technologies and sustainable chemical processes. The results presented in this account summarized the recent works of our group, which highlighted the "two birds one stone" strategies for green organic synthesis. In those cases, the reactant, catalyst or solvent may play dual roles rendering a cleaner organic synthesis process. In such procedures, the chemical waste and production costs were reduced, and the energy efficiencies were improved. In particular, the concept of dual roles design would open new routes for the development of green organic reactions.
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests:
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsWe are grateful for financial support from the National Natural Science Foundation of China (Nos. 21545010 and 31527803) and Hunan Provincial Natural Science Foundation of China (No. 2019JJ20008).
| [1] |
(a) S. Xu, Y. Zhou, J. Xu, H. Jiang, H. Liu, Green Chem. 15 (2013) 718-726; (b) L. Li, C. Shu, B. Zhou, et al., Chem. Sci. 5 (2014) 4057-4064; (c) D.Q. Dong, S.H. Hao, H. Zhang, Z.L. Wang, Chin. Chem. Lett. 28 (2017) 1597-1599; (d) L.X. Li, D.Q. Dong, S.H. Hao, Z.L. Wang, Tetrahedron Lett. 59 (2018) 1517-1520; (e) Y.H. Wang, G. Qiu, H. Zhou, W. Xie, J.B. Liu, Tetrahedron 75 (2019) 3850-3855; (f) Q. Sun, L. Liu, Y. Yang, Z. Zha, Z. Wang, Chin. Chem. Lett. 30 (2019) 1379-1382; (g) H. Yue, P. Bo, B.L. Wang, et al., Chin. J. Org. Chem. 39 (2019) 463-468; (h) T. Wei, Y. Zeng, W. He, L. Geng, L. Hong, Chin. Chem. Lett. 30 (2019) 383-385; (i) P. Bao, L. Wang, Q. Liu, et al., Tetrahedron Lett. 60 (2019) 214-218; (j) J. Li, W. Tang, D. Ren, J. Xu, Z. Yang, Green Chem. 21 (2019) 2088-2094; (k) K. Sun, S.J. Li, X.L. Chen, et al., Chem. Commun. 55 (2019) 2861-2864; (l) G. Qiu, Z.F. Chen, W. Xie, H. Zhou, Eur. J. Org. Chem. 2019 (2019) 4327-4333. |
| [2] |
(a) W.H. Bao, Z. Wang, X. Tang, et al., Chin. Chem. Lett. (2019), doi: http://dx.doi.org/10.1016/j.cclet.2019.06.052; (b) L.H. Lu, Z. Wang, W. Xia, et al., Chin. Chem. Lett. 30 (2019) 1237-1240; (c) J. Xu, W. Huang, R. Bai, et al., Green Chem. 21 (2019) 2061-2069; (d) L. Wang, M. Zhang, Y. Zhang, et al., Chin. Chem. Lett. (2019), doi: http://dx.doi.org/10.1016/j.cclet.2019.05.041. |
| [3] |
(a) S.B. Yu, H.J. Zang, X.L. Yang, et al., Chin. Chem. Lett. 28 (2017) 1479-1484; (b) C. Wu, L.H. Lu, A.Z. Peng, et al., Green Chem. 20 (2018) 3683-3688; (c) D.Q. Dong, W.J. Chen, Y. Yang, X. Gao, Z.L. Wang, ChemistrySelect 4 (2019) 2480-2483; (d) G.P. Yang, X. Wu, B. Yu, C. Hu, ACS Sustain. Chem. Eng. 7 (2019) 3727-3732; (e) M. Li, F. Wu, Y. Gu, Chin. J. Catal. 40 (2019) 1135-1140. |
| [4] |
(a) L. Peng, Z. Hu, Q. Lu, et al., Chin. Chem. Lett. (2019), doi: http://dx.doi.org/10.1016/j.cclet.2019.05.063; (b) M. Sun, J. Jiang, J. Chen, Q. Yang, X. Yu, Tetrahedron 75 (2019) 130456; (c) C.Wu, H.J.Xiao, S.W.Wang, etal., ACSSustain.Chem.Eng.7 (2019)2169-2175. |
| [5] |
(a) X. Gong, J. Chen, X. Li, W. Xie, J. Wu, Chem. -Asian J. 13 (2018) 2543-2548; (b) T.Y. Shang, L.H. Lu, Z. Cao, et al., Chem. Commun. 55 (2019) 5408-5419; (c) L.Y. Xie, T.G. Fang, J.X. Tan, et al., Green Chem. 21 (2019) 3858-3863; (d) S. Ye, T. Xiang, X. Li, J. Wu, Org. Chem. Front. 6 (2019) 2183-2199; (e) L.Y. Xie, L.L. Jiang, J.X. Tan, et al., ACS Sustain. Chem. Eng. 7 (2019) 14153-14160; (f) G.H. Li, D.Q. Dong, Q. Deng, S.Q. Yan, Z.L. Wang, Synthesis 51 (2019) 3313-3319; (g) S. Ye, X. Li, W. Xie, J. Wu, Asian J. Org. Chem. 8 (2019) 893-898; (h) D.Q. Dong, L.X. Li, G.H. Li, Q. Deng, Z.L. Wang, Chin. J. Catal. 40 (2019) 1494-1498; (i) X.C. Liu, K. Sun, X. Chen, et al., Adv. Synth. Catal. 361 (2019) 3712-3717. |
| [6] |
L.Y. Xie, Y.J. Li, J. Qu, et al., Green Chem. 19 (2017) 5642-5646. DOI:10.1039/C7GC02304A |
| [7] |
(a) Q. Huang, L. Zhu, D. Yi, X. Zhao, W. Wei, Chin. Chem. Lett. (2019), doi: http://dx.doi.org/10.1016/j.cclet.2019.07.049; (b) Z. Gan, Q. Yan, G. Li, et al., Adv. Synth. Catal. 361 (2019) 4558-4567; (c) X.M. Xu, D.M. Chen, Z.L. Wang, Chin. Chem. Lett. (2019), doi: http://dx.doi.org/10.1016/j.cclet.2019.05.048; (d) G. Li, Z. Gan, K. Kong, X. Dou, D. Yang, Adv. Synth. Catal. 361 (2019) 1808-1814; (e) X. Wang, C. Li, Y. Zhang, B. Zhang, K. Sun, Org. Biomol. Chem. (2019), doi: http://dx.doi.org/10.1039/C9OB01530B; (f) Y. Chen, H. Qi, N. Chen, et al., J. Org. Chem. 84 (2019) 9044-9050; (g) Y. Liu, X.L. Chen, K. Sun, et al., Org. Lett. 21 (2019) 4019-4024; (h) Q. Zhu, C. Liu, L. Zhou, et al., Biosens. Bioelectron. 140 (2019) 175-182; (i) C. Zhu, D. Wei, Y. Wu, et al., J. Alloys. Compd. 778 (2019) 731-740; (j) X.Y. Qin, L. He, J. Li, et al., Chem. Commun. 55 (2019) 3227-3230; (k) Z. Shen, C. Pi, X. Cui, Y. Wu, Chin. Chem. Lett. 30 (2019) 1374-1378; (l) S. Liu, K. Chen, W.J. Hao, et al., J. Org. Chem. 84 (2019) 1964-1971. |
| [8] |
(a) S. Ye, X. Li, W. Xie, J. Wu, Eur. J. Org. Chem. (2019), doi: http://dx.doi.org/10.1002/ejoc.201900396; (b) X. Gong, G. Li, Z. Gan, et al., Asian J. Org. Chem. 8 (2019) 1472-1478; (c) J. Zhang, X. Li, W. Xie, S. Ye, J. Wu, Org. Lett. 21 (2019) 4950-4954; (d) L. Wang, Y. Zhang, M. Zhang, et al., Tetrahedron Lett. 60 (2019) 1845-1848; (e) H. Jiang, X. Tang, Z. Xu, et al., Org. Biomol. Chem. 17 (2019) 2715-2720; (f) D.F. Jiang, J.Y. Hu, W.J. Hao, et al., Org. Chem. Front. 5 (2018) 189-196. |
| [9] |
W. Li, G. Yin, L. Huang, et al., Green Chem. 18 (2016) 4879-4883. DOI:10.1039/C6GC01196A |
| [10] |
Z. Yang, Z. Song, L. Jie, L. Wang, X. Cui, Chem. Commun. 55 (2019) 6094-6097. DOI:10.1039/C9CC02232E |
| [11] |
(a) K. Sun, S. Wang, R. Feng, et al., Org. Lett. 21 (2019) 2052-2055; (b) X.H. Ouyang, R.J. Song, J.H. Li, Chem. Asian J. 13 (2018) 2316-2332; (c) L. Zhang, L. Ma, H. Zhou, et al., Org. Lett. 20 (2018) 2407-2411. |
| [12] |
C. Wu, X. Xin, Z.M. Fu, et al., Green Chem. 19 (2017) 1983-1989. DOI:10.1039/C7GC00283A |
| [13] |
(a) F.L. Zeng, X. Chen, S.Q. He, et al., Org. Chem. Front. 6 (2019) 1476-1480; (b) J. Zhang, W. Xie, S. Ye, J. Wu, Org. Chem. Front. 6 (2019) 2254-2259; (c) G. Li, Q. Yan, X. Gong, X. Dou, D. Yang, ACS Sustain. Chem. Eng. 7 (2019) 14009-14015; (d) Q. Liu, L. Wang, H. Yue, et al., Green Chem. 21 (2019) 1609-1603; (e) Z. Cheng, W. Jin, C. Liu, Org. Chem. Front. 6 (2019) 841-845; (f) B. Liu, L. Cheng, P. Hu, et al., Chem. Commun. 55 (2019) 4817-4820; (g) P. Hu, M. Tan, L. Cheng, et al., Nat. Commun. 10 (2019) 2425. |
| [14] |
K.J. Liu, S. Jiang, L.H. Lu, et al., Green Chem. 20 (2018) 3038-3043. DOI:10.1039/C8GC00223A |
| [15] |
(a) G.P. Yang, N. Jiang, X.Q. Huang, B. Yu, C.W. Hu, Mol. Catal. 468 (2019) 80-85; (b) B. Yu, B. Zou, C.W. Hu, J. CO2 Util. 26 (2018) 314-322. |
| [16] |
(a) L. Wang, D. Xiong, L. Jie, C. Yu, X. Cui, Chin. Chem. Lett. 29 (2018) 907-910; (b) Y. Gu, W. Huang, S. Chen, X. Wang, Org. Lett. 20 (2018) 4285-4289; (c) W. Wei, L. Wang, H. Yue, et al., ACS Sustain. Chem. Eng. 6 (2018) 17252-17257; (d) S.H. Hao, L.X. Li, D.Q. Dong, Z.L. Wang, X.Y. Yu, Tetrahedron Lett. 59 (2018) 4073-4075; (e) W. Xie, Y. Wu, J. Zhang, et al., Eur. J. Med. Chem. 145 (2018) 35-40; (f) K. Chen, W.J. Hao, S.J. Tu, B. Jiang, Green Chem. 21 (2019) 675-683; (g) Y. Liu, L. Chen, Z. Wang, et al., J. Org. Chem. 84 (2019) 204-215; (h) B. Wu, Z. Yang, H. Zhang, L. Wang, X. Cui, Chem. Commun. 55 (2019) 4190-4193; (i) S.Y. Wu, W.L. Chen, X.P. Ma, et al., Adv. Synth. Catal. 361 (2019) 965-970; (j) Z. Yang, L. Jie, Z. Yao, Z. Yang, X. Cui, Adv. Synth. Catal. 361 (2019) 214-218; (k) G.H. Li, D.Q. Dong, Y. Yang, X.Y. Yu, Z.L. Wang, Adv. Synth. Catal. 361 (2019) 832-835; (l) G.H. Li, D.Q. Dong, X.Y. Yu, Z.L. Wang, New J. Chem. 43 (2019) 1667-1670; (m) M. Li, X. Dong, N. Zhang, F. Jérôme, Y. Gu, Green Chem. 21 (2019) 4650-4655; (n) J. Wang, K. Sun, X. Chen, et al., Org. Lett. 21 (2019) 1863-1867; (o) Y. Zhang, K. Sun, Q. Lv, et al., Chin. Chem. Lett. 30 (2019) 1361-1368; (p) L. Xu, T. Li, L. Wang, X. Cui, J. Org. Chem. 84 (2019) 560-567. |
| [17] |
(a) Y. Pan, G.W. Chen, C.H. Shen, W. He, L.W. Ye, Org. Chem. Front. 3 (2016) 491-495; (b) W.B. Shen, Q. Sun, L. Li, et al., Nat. Commun. 8 (2017) 1748; (c) R. Zhang, S. Luo, Chin. Chem. Lett. 29 (2018) 1193-1200; (d) Q. Liang, Y. Zhang, M. Huang, Y. Xiao, F. Xiao, Mol. Med. Rep.19 (2019) 1256-1265; (e) Y. Zhang, Y. Zhang, Y. Xiao, C. Zhong, F. Xiao, Ecotoxicol. Environ. Saf. (2019), doi: http://dx.doi.org/10.1016/j.ecoenv.2019.109465. |
| [18] |
J. Wu, X. Cui, L. Chen, G. Jiang, Y. Wu, J. Am. Chem. Soc. 131 (2009) 13888-13889. DOI:10.1021/ja902762a |
| [19] |
J.W. Yuan, L.B. Qu, Chin. Chem. Lett. 28 (2017) 981-985. DOI:10.1016/j.cclet.2017.01.016 |
| [20] |
(a) K. Sun, X.L. Chen, X. Li, et al., Chem. Commun. 51 (2015) 12111-12114; (b) Z. Wu, H. Song, X. Cui, et al., Org. Lett. 15 (2013) 1270-1273. |
| [21] |
B. Du, P. Qian, Y. Wang, et al., Org. Lett. 18 (2016) 4144-4147. DOI:10.1021/acs.orglett.6b02289 |
| [22] |
L.Y. Xie, S. Peng, F. Liu, et al., Org. Chem. Front. 5 (2018) 2604-2609. DOI:10.1039/C8QO00661J |
| [23] |
L.Y. Xie, S. Peng, F. Liu, et al., Adv. Synth. Catal. 360 (2018) 4259-4264. DOI:10.1002/adsc.201800918 |
| [24] |
J. Sun, Y. Li, Y. Gui, et al., Chin. Chem. Lett. 30 (2019) 569-572. DOI:10.1016/j.cclet.2018.11.024 |
| [25] |
L.Y. Xie, J. Qu, S. Peng, et al., Green Chem. 20 (2018) 760-764. DOI:10.1039/C7GC03106H |
| [26] |
L.Y. Xie, S. Peng, T.G. Fan, et al., Sci. China Chem. 62 (2019) 460-464. DOI:10.1007/s11426-018-9446-1 |
| [27] |
(a) X. Wang, M. Yang, W. Xie, X. Fan, J. Wu, Chem. Commun. 55 (2019) 6010-6013; (b) K. Sun, Z. Shi, Z. Liu, et al., Org. Lett. 20 (2018) 6687-6690; (c) X. Gong, X. Li, W. Xie, J. Wu, S. Ye, Org. Chem. Front. 6 (2019) 1863-1867; (d) F.L. Zeng, X.L. Chen, S.Q. He, et al., Org. Chem. Front. 6 (2019) 1476-1480; (e) Y. Zong, Y. Lang, M. Yang, et al., Org. Lett. 21 (2019) 1935-1938; (f) K. Sun, B. Luan, Z. Liu, et al., Org. Biomol. Chem. 17 (2019) 4208-4211; (g) C. Pi, X. Yin, X. Cui, Y. Ma, Y. Wu, Org. Lett. 21 (2019) 2081-2084; (h) P. Bao, H. Yue, N. Meng, et al., Org. Lett. 21 (2019) 7218-7222. |
| [28] |
L.H. Lu, S.J. Zhou, M. Sun, et al., ACS Sustain. Chem. Eng. 7 (2019) 1574-1579. DOI:10.1021/acssuschemeng.8b05344 |