 2019, Vol. 30
2019, Vol. 30 


b National Engineering Research Center for Flue Gas Desulfurization, Sichuan University, Chengdu 610065, China
In recent years, with the rapid development of the global economy, the air haze pollution problems have become increasingly prominent in China, which principal substance is PM2.5 [1]. PM2.5 refers to atmospheric fine particles with an aerodynamics diameter less than 2.5 μm. According to the Global Burden of Disease, environmental pollutionfrom PM2.5 accountedfor 3.1 milliondeaths worldwide as of 2010 [2]. Moreover, the transportation distance of haze is verylong, whichwill reduce the visibility of a large area of the city and greatly reduce the effective visual distance [3-5]. The main components of PM2.5 are secondary particles such as sulfate, nitrate and ammonium salt, and their contribution is as high as 50%, which are mainly produced by the chemical transformation of acidic gases such as sulfur dioxide (SO2), nitrogen oxide (NOx) and hydrogen sulfide (H2S) in the atmosphere [6, 7]. In addition, global warming caused by massive emissions of greenhouse gases such as carbon dioxide (CO2) is another major environmental problem. It causes temperature rise, sea level rise, land desertification, increased disease and insect pests and so on [8].
These acidic gases such as SO2, NOx, H2S and CO2, are mainly emitted from anthropogenic sources. The total emissions of SO2 in China accounts for about 26% of the global emissions, which is over 19 million tons per year for more than ten years, and about 85% comes from the combustion of coal-based fossil fuels [9]. The NOx are mainly derived from fossil fuel combustion, metal smelting, municipal solid waste incineration and so on, and NOx emissions of China has increased to 25 million tons in 2012 [10]. The main sources of H2S are natural gas processing, petroleum refinement, gas manufacturing and wastewater treatment. The CO2 derived mainly from fossil fuel combustion and automobile exhaust emissions, and it is estimated that the total CO2 emission is responsible for 2/3 of the growth of all greenhouse gas emissions [11]. Thus, it is significantly important to take effective measures for the control of the emission of these gases into atmosphere.
At present, the commonly used purification methods of gaseous pollutants are absorption, adsorption, catalytic and biological methods. Among them, adsorption method is one of the most widely used methods, because of the high selectivity, low cost, good separation effect and high purification efficiency [12]. Adsorption is divided into physical adsorption and chemical adsorption, based on the intermolecular force and chemical bond of adsorbents and adsorbates, respectively. The adsorbents commonly used can be divided into metals and their compounds such as MnO2 and nonmetallic materials such as carbon-based materials. Supported metal compounds can be used as the adsorbents for the removal of SO2, NOx, H2S and CO2, i.e., Fe, Co, Ni, Cu, Cr, Ce [13-17]. Jin et al. [16] used pyrolusite slurry to absorb SO2 from industrial waste gases and found that the removal rate of SO2 can reach more than 90%. Ren et al. [17] found that the removal rate of SO2 was nearly 100% and the leaching rate of manganese was close to 100% using a self-developed jet bubbling reactor. On the other hand, there are mainly two kinds of nonmetallic acidic gases adsorbents, including activated carbon [18-20] and zeolite molecular sieve [21-26]. Mochida et al. [24] believed that SO2, O2 and H2O molecules are adsorbed firstly on the carbon surface, while the formation of H2SO4 can be achieved, when there is a close enough distance and the appropriate spatial configuration between them. Zeolite molecular sieve can also adsorb SO2 effectively because of its good porosity and permeability [25, 26]. However, from the experimental operation, the various reactions can only be judged from the macroscopic change of pollutants substances, while the specific mechanism of adsorption reactions cannot be explained very well.
First-principles calculation is valuable in understanding surface adsorption reactions, adsorption reactivity, and structure-activity relationships [27-31]. The first-principles calculation is an algorithm for solving the Schrödinger equation directly after some approximate treatment according to the principle of the interaction between nucleus and electron, and its basic motion law, using the principle of quantum mechanics, starting from the specific requirements [32]. In recent years, with the vast development of various algorithms and high-performance computing equipment, first-principles calculation have been widely used in various surface chemical reactions, including surface adsorption reactions [33, 34], which provides reliable theoretical guidance for practical applications and makes up for some deficiencies in experimental operation. Especially in the field of adsorption, it is possible to understand the energy change, charge density and magnetic properties of pollutant molecules adsorbed on different materials by the method of first-principles calculation [35, 36], which can help us to explain the experimental results more effectively in terms of mechanism.
Thus, this review first introduced the theoretical basis behind first-principles calculation, then some commonly software, and application examples of acidic gases (i.e., SO2, NOx, H2S and CO2) adsorbed on metal and nonmetallic surfaces studied by first-principles calculation are summarized. This review will provide theoretical basis for further modification of catalysts for removing these polluting gases, so as to further improve their removal efficiency, reduce energy consumption and prolong the lifetime of catalysts. It also helps to encourage researchers in this field to consider first-principles calculations when studying surface reactions at the atomic scale.
2. Theory section 2.1. OverviewQuantum mechanics is a physical theory that describes microscopic matter. The quantum mechanics of matrix expression are established by W. K. Heisenberg in 1925 [37]. The following year, the classical wave equation is proposed by E. Schrödinger and it is discussed that the equation is essentially unified with the expression of quantum mechanics matrix [38]. The generation of quantum mechanics provides a way to describe the motion of electrons and nuclei under arbitrary conditions [39, 40]. All properties of the material can be obtained by solving the Schrödinger equation in principle. In the microscopic field, the movement of electrons determines most of the properties of the material, but the interaction between electrons in the material is highly complex. Moreover, in multi-particle research systems such as gas phase molecules, clusters, and crystals, the interaction between particles is extremely complicated. Therefore, it is difficult to understand the properties of multibody materials strictly based on the solution of Schrödinger equation. The use of density functional theory (DFT) can solve this problem well and it is more effective to investigate the properties of the materials.
2.2. Density functional theoryIn the 1960s, the DFT was proposed by P. Hohenberg, W. Kohn and Sham [41, 42]. DFT is a quantum theory based on Thomas-Fermi's theory, in which the electron density distribution n(r) is used as the basic variable to study the electronic structure of atoms and molecules. In the traditional quantum theory system, wave function is regarded as the basic physical quantity. However, the electron density is regarded as the basic variable in DFT calculation. Hence, the solution of Schrödinger equation is transformed into the solution of the optimal electron density based on the DFT. Meanwhile, the multi-electron problem is successfully transformed into the single-electron equation [43, 44]. In recent years, the combination of DFT and molecular dynamics has made a lot of progress in material calculation, and has gradually become the core technology in materials science and related fields.
Hohenberg and Kohn put forward the basic theoretical basis of DFT, which is called Hohenberg-Kohn theorem [45, 46]. Although the Hohenberg-Kohn theorem establishes the framework of DFT, it has not explained how to establish its concrete form. Therefore, it cannot be directly used to solve the problem. In 1965, Kohn worked with Sham to introduce a multi-electron system with no interaction in the potential field. The charge density of the system is the same as that of the original system, and the kinetic energy of the non-interacting system can be simply written as the sum of each electron kinetic energy. With these basic theoretical frameworks and dealing with the exchange correlation energy, the Schrödinger equation of the ground state of the complex multielectron system can be transformed into a simple single-electron equation, which brings great convenience to scientific research. However, they do not accurately express the exchange correlation energy, so it is still difficult to get practical application. In order to solve this problem, the local density approximation (LDA) was proposed after the Kohn-Sham equation was put forward in the same year [46]. LDA assumes that the functional of charge density of the system changes slowly in space, so the charge density in each volume element is affected only by its spatial position. This approximate method can accurately find the exchange correlation functional and is suitable for calculating the uniform distribution of charge density in space. However, the LDA approximation has a great error in the calculation of transition metals in systems where the charge density varies more sharply. For the sake of obtain more accurate results in different calculation processes, the inhomogeneity of charge distribution must also be considered. To solve this problem, Becke introduced the gradient of charge density in 1988 to modify the result of LDA calculation, which resulted in the generalized gradient approximation (GGA) [47]. The method assumes that the exchange correlation energy is related to the local charge density and the nearby charge density and can deal with the non-uniform system, so the calculated results are more accurate. At present, the GGA method has developed a variety of models, such as the common Perdew-Burke-Emzetho (PBE) [48], Perdew Wang 91 (PW91) [49], Becke and BLYP exchange correlation potential. At present, in order to improve the accuracy of theoretical calculation, the hybrid functional method is introduced, that is, the exchange correlation functional of the system is obtained by using the exchange energy of Hartree-Fock and the exchange energy in DFT as a linear combination. Such as the commonly used HSE06 hybrid functionals and B3LYP functionals in Gaussian calculation method [50].
2.3. Software packages based on DFTWith the above theoretical foundation, DFT has been continuously developed and widely used in practice. In order to satisfy the huge computational requirements of first-principles calculation, scientists have developed various computing programs and software after combining with the modern computer technology through continuous research and experiments. This software can be used on a variety of large computers, servers and personal computers with a wider range of applications and better compatibility. And these programs and software have different features and can be applied to different computing systems. Therefore, researchers can choose the software according to their actual demand in practical application. Here, we will introduce several kinds of software commonly used by researchers: VASP, Material Studio and Gaussian.
The VASP (Vienna Ab-initio Simulation Package) is a commercial software package to describe the electronic-core interaction used in quantum mechanical and molecular dynamics calculation, which comes from the Hafner team of the University of Vienna [51, 52]. It is one of the most popular software in the related fields of science [53-55]. VASP used plane wave pseudopotential method to carry out ab initio simulation, and used periodic boundary conditions to deal with atoms, molecules, clusters, crystals, thin films, solids and surface systems, etc. The electronic structure, optical and magnetic properties of the crystal can be calculated more exactly. Similarly, some basic physical and chemical properties of the material surface can also be calculated accurately. VASP has been improved step by step as the continuous development of computer science and technology. Now VASP has been widely used in the optimization of complex systems and the adsorption of molecules at the surface, and can be used for high efficiency parallel computation of mainframe computers.
Material Studio is a powerful platform for material science researchers, and it is also a powerful modeling tool for configuration optimization, property prediction, X-ray diffraction analysis and the calculation of complex dynamic simulation. Material Studio contains several modules to perform varies calculations, including Materials Visualizer, Discover, COMPASS, Amorphous Cell, Reflex, DMol3, CASTEP and other modules. Among them, DMol3 and CASTEP are widely applied in various areas. Unlike other quantum mechanical programs, DMol3 uses numerical functions to describe atomic orbitals. The method describing the electronic state of the system is the linear combination of atomic orbitals, which results in extremely high computational accuracy and efficiency. DMol3 can also simulate the chemical reaction processes of gaseous, liquid, and solid surfaces, which is one of its major characteristics. Therefore, it has been widely applied in the fields of physics, chemistry, biology, chemical engineering, in particular to the field of catalytic reaction and other molecular reactions. CASTEP uses the plane wave pseudopotentials to carry out the first-principles calculation. It is often used to study metals, semiconductors, ceramics, and so on. The surface chemical and optical properties of crystals and the wave functions of the system can be obtained. Now it has been widely used in solid state physics, material science and other fields.
Gaussian is also a comprehensive software package widely used in the field of quantum chemistry. It can be applied not only to density functional method, but also to molecular dynamics, semiempirical method and so on. Gaussian is often used to calculate molecular orbitals, vibrational frequencies, thermodynamic properties, FT-IR and Raman spectra, multiple moment, reaction paths, etc. The calculation can be performed on the ground state or excited state of the system and predict the energy, structure and molecular orbital of the periodic system. It is suitable for the study of chemical reaction systems [56, 57].
3. ApplicationsThe adsorption properties of several acidic gaseous pollutants (SO2, NOx, H2S and CO2) on different materials are discussed in this section. Adsorption materials we discussed mainly include metals and their compounds, and non-metallic materials, such as graphene and nanotubes. DFT method is used to calculate the adsorption process and properties of these substances at different interfaces. Some representative examples are selected to discuss the application of first-principles calculation to the adsorption of these pollutants.
3.1. Adsorption of acidic gas molecules on metal surfaceTransition metals and precious metals play a significant role in the catalytic oxidation of some gas pollutant molecules such as SO2 and NOx, and often exist as catalysts with excellent performance [58-66]. In general, the experimental study is to make metals into large single crystals. After cutting the single crystals, the required crystal faces can be obtained, which provides a material basis for the adsorption of the latter molecules. The lattice of the metal is replaced by different forms of crystal planes, which are represented by Miller indices. For example, the metal Fe has (100), (110) and (111) plane. At the same time, certain metal oxides may have some properties which are not found in single metal, such as copper oxides have higher sulfur capacity and higher affinity to SO2 molecules as desulfurizer [67]. From the last century, some scholars have studied the adsorption behavior of gas molecules on some metal surfaces by DFT theories [68-72]. Therefore, the study of the adsorption or decomposition mechanism of these pollutants on the surface of metal and its compounds is of great significance for improving the existing catalysts and producing new catalysts.
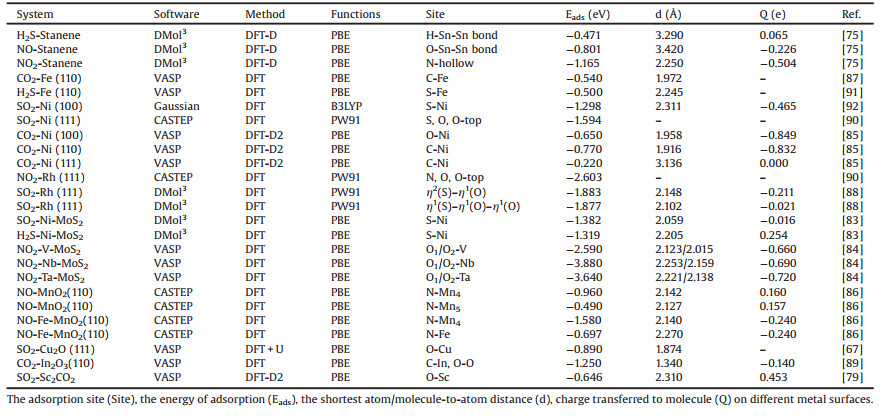
The metal monolayer-stanene material is a newly discovered zero-gap semiconductors with buckled honeycomb structure, which has attracted extensive attention in material science and other disciplines [73, 74]. Chen Xianping et al. [75] studied the adsorption properties of H2S, NO, NO2 and other gas molecules on the surface of stanene by first-principles calculations. There are three absorbable positions in stanene, which are the top of Sn atom, the center of a Sn hexagonal and the center of Sn-Sn bond. It is found that the interaction of H2S and stanene is weak and belongs to physical adsorption. There is a strong interaction between NO and stanene, and the adsorption energy is -0.801 eV. However, the adsorption energy of NO2 on stannene is above -1.12 eV, and its configuration is shown in Fig. 1. At the same time, it is found from density of states (DOS) that the Sn p and O p orbitals of the NO2/stanene structure have strong orbital hybridization, which shows that the adsorption structures for NO2 adsorbed on stanine are quite stable. And the charge density difference (CDD) also proved this result. The results indicate that stanene has a potential application in the fields of gas sensor and high-performance catalyst for the detection and adsorption of NO and NO2.

|
Download:
|
| Fig. 1. Top and side view of the lowest energy structure of NO2 adsorbed on stanene. Reproduced with permission [75]. Copyright 2016, The Journal of Physical Chemistry C. | |
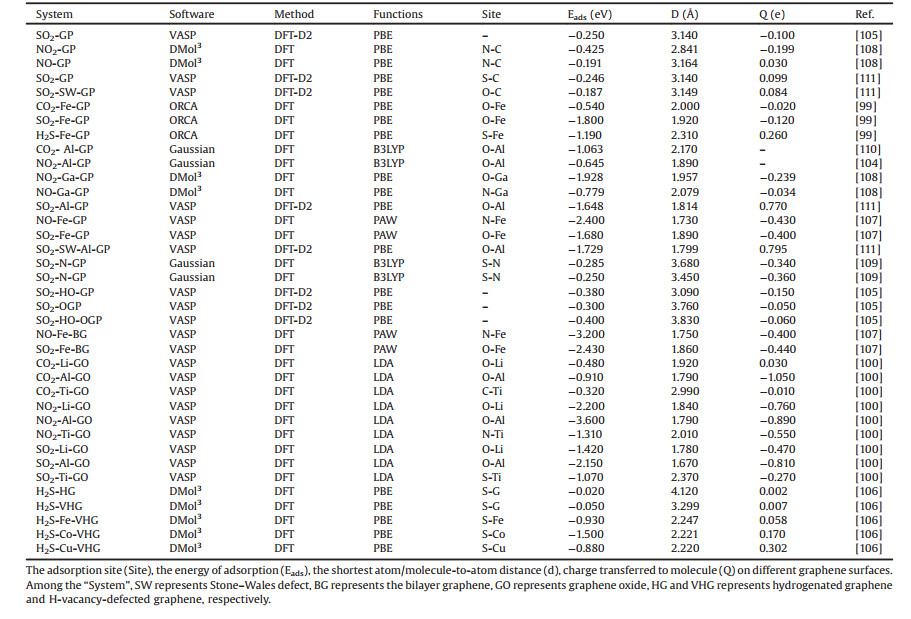
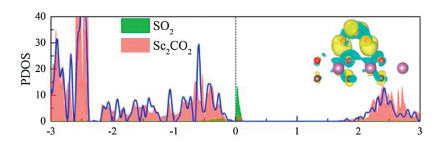
In recent years, some oxygen-functionalized MXenes materials, i.e., M2CO2 (M = Sc, Hf, Zr, Ti), have been found to have a broad prospect in gas molecular sensing or capture owing to their semiconductor characters [76-78]. Ma Shuhong et al. [79] have calculated certain properties of SO2 gas molecules adsorption on M2CO2 (M = Sc, Hf, Zr, Ti). By finding the favorable configuration of M2CO2 and the most stable structure of SO2 adsorbed on the surface of the material, it is found that Sc2CO2 is the most preferred for the adsorption of SO2 molecules. Its adsorption energy is -0.646 eV. As seen from Fig. 2, the significant orbital hybridizations between SO2 and Sc2CO2 are localized near the Fermi level, so that SO2 adsorbs on Sc2CO2. Therefore, the charge transfer between the SO2 and Sc2CO2 increases multiply, which is consistent with the CDD diagram on the right of Fig. 2, indicating that the adsorption is relative stable. Moreover, the method of improving the adsorption capacity of SO2 by applying tensile strains or negative electric fields has been proposed. All these results show that Sc2CO2 can be considered as a gas sensor or adsorption material for SO2.

|
Download:
|
| Fig. 2. PDOS of SO2 molecule (green shadow), Sc2CO2 monolayers with (red shadow) and without (blue solid line) SO2 adsorption, respectively. The side views of charge density difference are given in the upright panels. Reproduced with permission [79]. Copyright 2017, The Journal of Physical Chemistry C. | |
MoS2 is a new type of transition metal 2D material with excellent structure and properties, such as high activity, high selectivity and high stability, now it has attracted extensive attention in the fields of chemistry [80-82]. On the basis of the first-principle calculations, Wei Huangli et al. [83] studied the adsorption properties of the original MoS2 monolayer and the molecular layer doped with Ni metal atoms to the H2S and SO2 gas molecules, and compared the adsorbed structural parameters, the adsorption energy and the amount of charge transfer. The adsorption energy of H2S and SO2 reaches -1.319 eV and -1.382 eV, respectively, indicating that the interaction between the two gases and Ni-doped MoS2 monolayers is pretty strong. The doping of metal atoms provides a substantial number of free electrons for MoS2, and the strong orbital hybridization with adsorbed molecules improves the adsorption capacity of the MoS2 molecular layer. Accordingly, Ni-doped MoS2 might be a promising gas absorbent material to remove H2S and SO2 gas pollutants.
Table 1 summarizes the adsorption examples of acidic gas pollutant molecules on different metals and their compounds, and gives the software, methods and results of adsorption [67, 75, 79, 83-92]. According to the reported results, gas molecules may be adsorbed on the surface of materials in different states, and there are many adsorption sites on the surface of materials. Therefore, the adsorption configurations of these gas molecules on different surfaces of materials are not one way but many ways. In order to obtain the desired results briefly, the most stable configuration of adsorption and the corresponding results are selected. It can be seen from Table 1 that the adsorption energy of the gas molecules is relatively large, which belongs to the chemical adsorption process. Moreover, we found that the adsorbent materials are mainly transition metal elements. In addition, doping another transition metal element into metal compounds can significantly improve the adsorption properties of the materials [83, 86]. These results indicate that these acidic gas pollutant molecules could undergo a great adsorption reaction on the transition metal and its compounds.
|
|
Table 1 Summary of adsorption of acidic gas molecules on metal surface. |
3.2. Adsorption of acidic gas molecules on non-metallic surface 3.2.1. Adsorption of gas molecules on graphene surface
Graphene (GP) is a kind of two-dimensional monatomic carbon simple material with perfect hexagonal structure [93]. It is also a multifunctional catalyst carrier with high conductivity, high structure stability and high mechanical strength. Its structure is a two-dimensional crystal structure composed of hexagonal crystals. There are two kinds of carbon atoms in each unit cell. After oxidation with strong oxidant, various graphene oxides can be obtained, and it is easy to be modified. At the same time, it can form a stable structure with other atoms to carry out a series of reaction processes. For example, the chemical activity of the system can be enhanced by decoration of graphene with adatoms or dopants, and the bonding strength of gases on graphene can be improved at the same time [94-98]. Therefore, graphene has been widely used in chemical catalysis, adsorption and other related fields.
Based on DFT, Diego Cortés-Arriagada et al. [99] calculated the adsorption behavior of CO2, SO2 and H2S and other pollutant gases on graphene nanosheets doped with Fe atoms. The adsorption capacity of the original graphene and the doped graphene for gas molecules was compared. The adsorption of H2S onto pristine graphene and Fe-doped graphene is shown in Fig. 3. The results show that the adsorption energy of CO2, SO2 and H2S gas on graphene are about -0.11, -0.28 and -0.15 eV, respectively, while the adsorption energy on Fe-GP is about -0.54, -1.80 and -1.19 eV.

|
Download:
|
| Fig. 3. The adsorption of H2S onto pristine graphene (G) and Fe-doped graphene (FeG). Reproduced with permission [99]. Copyright 2018, Applied Surface Science. | |
The chemical adsorption is still strong even at room temperature. It is demonstrated that graphene doped with Fe atom is a feasible catalyst or absorbent. Chen Chi et al. [100] reported the adsorption of CO2, NO2, SO2 on metal (Li, Al, Ti) modified graphene oxide (GO) by VASP by using the first-principles calculations method. Hydroxyl and epoxy groups were placed on the hexatomic ring to obtain the most stable configurations of metal decorated GO. It is found that the binding energies of CO2, NO2 and SO2 on Li, Al, Ti decorated GO surface are about -0.19 to -0.91 eV, -1.25 to -3.60 eV and -0.44 to -2.15 eV, respectively. However, other scholars have studied that the binding energies of CO2, NO2 and SO2 on graphene surface are -0.40 [101], 0.07 [102] and 0.01 [103] eV, respectively. In addition, the adsorption distance between gas molecules and GO surface and the results of Bader charge analysis are consistent with the results of binding energies, as shown in Fig. 4, an example is given. The results show that metal decorated GO has a significant effect on the capture of CO2, NO2 and SO2 gases.

|
Download:
|
| Fig. 4. The bond angles (green) of acidic gases (a is CO2; b is NO2; c is SO2), the distances (black) from gases to metals and the electron transfers (blue) between gases and metal decorated GO (a–c is Li decorations). Reproduced with permission [100]. Copyright 2014, Physical Chemistry Chemical Physics. | |
Ali Shokuhi Rad [104] also used the first-principles calculation method to study the adsorption properties of NO2 on Al-doped graphene. In the pristine graphene sheets, one C atom was substituted by one Al atom to form a stable structure. The results show that the adsorption energy of NO2 molecules on origin graphene is about -0.002 eV, while the NO2 have the lager adsorption energy on the Al-doped graphene of -0.645 eV, demonstrating that the presence of Al increases the capability of graphene to adsorb these NO2 molecules. The results of charge transfer and other properties also illustrate that doping metal atoms can significantly improve the interaction between adsorbates and substrate. Therefore, Al-doped graphene material is expected to be an excellent gas sensor material or adsorption material for NO2.
The adsorption properties of SO2 on the surface of graphene doped with three groups was calculated by Zhang Huijuan et al. [105] with van der Waals force modified DFT method. The first was doped with hydroxyl group, the second was doped with epoxy group, and the third was doped with both hydroxyl and epoxy group. Three kinds of graphene oxides (GO) were obtained. The adsorption structure, adsorption energy and charge transfer were calculated and compared with the adsorption of SO2 on graphene. The results reveal that the metallicity of GO decreases with the addition of oxygen functional groups, which leads to the decrease of SO2 adsorption by GO. However, the adsorption energy of SO2 on graphene surface doped with hydroxyl group is larger than pristine graphene because of the formed hydrogen bond between hydroxyl group and SO2. Through the Bader charge analysis and differential charge density analysis, it can be found that the existence of hydroxyl group can promote the charge transfer, while the existence of epoxy group will hinder the charge transfer. It is concluded that GO doped with hydroxyl groups can enhance the adsorption of SO2.
Table 2 summarizes the adsorption examples of these acidic gas pollutant molecules on different graphene, and gives the software, methods and results of adsorption [99, 100, 104-111]. The most stable configuration of adsorption and its corresponding calculation results are still selected. We can see from the results in Table 2 that the pure graphene is different from the structurally modified graphene. Although the gas adsorbed is different, they all have a relatively similar rule: (ⅰ) The adsorption energy between pure graphene and gas molecules is small and belongs to weak physical adsorption, while the adsorption distance between the molecule and the surface of graphene is longer and the charge transfer amount is less. Therefore, their adsorption is extremely unstable, while the structure modified graphene often adsorbs these gas molecules by chemisorption. (ⅱ) The adsorption energy of modified graphene is larger than that of the pristine graphene and the adsorption distance is closer. The charge transfer is also changed to a certain extent. Therefore, they are easy to form stable adsorption structure. According to the results, we found that the adsorption performance of graphene doped with transition metal atoms on gas molecules was greatly improved in the modified graphene [106, 108, 111]. This indicates that doped groups or transition metal atoms in graphene is favorable to gas adsorption compared with the original structure of graphene, which is vastly different from the adsorption properties on the metal surface.
|
|
Table 2 Summary of the adsorption of acidic gas molecules on graphene surface. |
3.2.2. Adsorption of gas molecules on nanotubes surface
Since the discovery of carbon nanotubes (CNTs) in 1991 [112], scientists have found that CNTs have many special properties, such as great strength and elasticity, nanoscale space. CNTs are essentially graphene sheets rolled into seamless tubular monolayers, and composed of carbon atoms on the surface [113]. According to the different surface shapes of carbon atoms, they can be divided into armchair type, sawtooth type and spiral type. They have good bonding properties and have greater reaction activity than other graphite. The pore structure on the surface and inside of carbon nanotubes determines that it can be used as a good gas adsorbent and has a strong adsorption potential energy for some gases. The properties of BN are similar to that of C, and carbon nanotubes and fullerenes may be formed when there are layered structures in bulk structures in theory. Therefore, the BN layer was curled up like C layer to form BN nanotubes (BNNTs), and the structural properties of BNNTs were studied. Consequently, using carbon nanotubes and BNNTs as a new gas adsorbent candidate and studying their adsorption mechanism theoretically are of great significance for breaking through the traditional adsorption theory and methods.
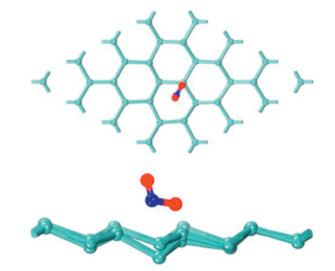
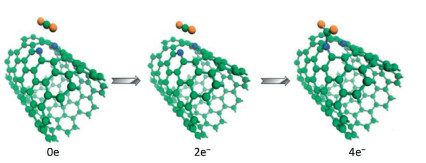
Based on DFT calculations, the sensing and adsorption properties of NO2 gas molecules on single-wall carbon nanotubes (SWCNTs) doped with metal atoms were calculated by Icell M. Sharafeldin et al. [114]. The (8, 0) SWCNT was selected as the matrix material, and a single carbon atom was replaced with a Cu, Pt or Ti to form a stable nanotube structure. The adsorption energy of NO2 on their surface come to reach -1.827 eV, -2.262 eV and -2.653 eV, respectively. However, it has been reported that the adsorption energy of NO2 on the surface of pristine CNTs is only about -0.267 eV [115], which indicates that there are strong interactions between metal doped SWCNTs and NO2 gas molecules. These conclusions prove that SWCNTs doped with Cu, Pt or Ti materials has high sensitivity to NO2 molecules. Jiao Yan et al. [116]. calculated the adsorption behavior of (7, 7) SWCNTs for CO2 under several nitrogen doping conditions, including graphite nitrogen, pyridine nitrogen and tripyridine nitrogen. The results show that the adsorption energy of CO2 molecules by all structures is less than 0.3 eV. However, when four extra electrons are injected into CNT doped with pyridine nitrogen, CO2 molecules can be adsorbed spontaneously by forming C–N bonds, and the adsorption capacity is significantly improved. Fig. 5 shows the adsorption configurations with 0, 2, and 4 electrons injection. According to this, the feasibility of electrocatalytic in gas adsorption and separation is considered, and a new thought for the preparation of gas adsorbents is provided.

|
Download:
|
| Fig. 5. Adsorption configurations of CO2 on pyridine-CNT with 0, 2, and 4 electrons injection. Reproduced with permission [116]. Copyright 2014, ChemSusChem. | |
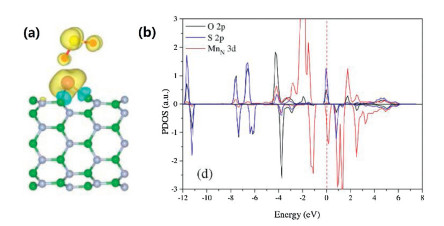
The adsorption behavior of SO2 molecules on BNNTs surface of original and doped with Mn atom was studied by Deng Zunyi et al. [117]. The simulated model uses zigzag (8, 0) BNNT to form two different Mn-BNNT configurations by replace one B atom or N atom with a Mn atom. The formation energy of SO2 molecule by pristine BNNT is only about -0.056 eV, which indicates that SO2 undergoes weakly physical adsorption. However, the formation energy of B and N sites of SO2 on Mn-BNNT is about -0.993 eV and -1.569 eV, which is much stronger than the original ones. As can be seen from Fig. 6, there is a strong covalent interaction and charge transfer between SO2 and MnN-BNNT. Therefore, the doping of Mn improves the adsorption capacity of BNNT remarkably, and provides a theoretical basis for the further application of BNNTs.

|
Download:
|
| Fig. 6. SO2 adsorbed on MnN-BNNT site. (a) Difference spin charge density of side view. (b) PDOS. Reproduced with permission [117]. Copyright 2015, Applied Surface Science. | |
Table 3 summarizes the adsorption examples of common acidic gaseous pollutant molecules on the surface of nanotubes, and gives the software, methods and results of adsorption [36, 114-121]. The most stable configuration of adsorption and its corresponding calculation results are still selected. According to the results of literature, we find that the adsorption ability of gas molecules on the surface of carbon nanotubes is weak and belongs to physical adsorption. However, on the surface of carbon nanotubes doped with metal atoms, the adsorption ability of gas molecules is relative stronger, and the adsorption structure is more stable. This is similar to the adsorption results of graphene. Similarly, we can conclude that the doping of metal atoms facilitates the adsorption of gas molecules on carbon nanotubes. It also shows that carbon nanotubes are theoretically feasible as adsorbents for acidic gaseous pollutant molecules.
|
|
Table 3 Summary of the adsorption of acidic gas molecules on nanotubes surface. |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
4. Conclusions and prospects
The first-principles calculation, especially the DFT method, is extremely useful for studying the adsorption process of gases on different materials. In this paper, we summarize the adsorption properties of several common acidic pollutants in the atmosphere on the surface of several kinds of materials. It was discussed on the calculated results of adsorption energy, the change of bond length and the configuration before and after adsorption, and the charge transfer of adsorption. The results suggested that SO2, NOx, H2S and CO2 can be adsorbed on metal and its compounds, graphene and nanotubes. Among them, metals and its compounds can achieve good adsorption effect without doping. The adsorption effect of the original graphene and carbon nanotubes is weak, while the adsorption performance can be greatly improved by doping the functional groups or metal atoms, especially the transition metal atoms. Accordingly, it is concluded that doping transition metal atoms in these adsorption materials is beneficial to the gas adsorption process. In addition, the first principles can investigate more properties of molecular adsorption, such as bond angle, energy band, density of state, magnetic properties, which have not been summarized in this paper. Through the calculation of these properties, we can obtain a series of physical and chemical change between them, which can explain some of the results obtained in the experimental research and provide theoretical support for the actual research results. Therefore, this is a significant and valuable work.
Although the first-principles calculation has been widely used in adsorption in recent years, we still put forward some prospects and suggestions: ⅰ) Metal and its compounds and graphene have been used to adsorb SO2, NOx and other gas molecules, which have been widely used in experimental research and some applications in practical engineering. However, the application of nanotubes in gas adsorption is still very few, and most of them are used in gas sensors. Hence, we hope that this review will give some inspiration to the researchers in the study of new adsorption materials. ⅱ) According to the summary of this paper, it can be found that noble metal elements have very good effect on the adsorption of several gas molecules, but the cost of using these precious metal materials in the actual experiment is relatively high. Therefore, we hope that this review can be used as a theoretical reference for researchers to study the adsorption of related gas molecules on related materials, so as to develop more excellent adsorbents with good performance, and even develop more economical and green adsorption materials that can replace precious metal elements. ⅲ) The first-principles calculation often has ideal conditions for simulating the molecular and material surfaces, and the adsorption materials used in the actual experiments are not pure substances. In addition, the experimental conditions are changeable and cannot be simulated, so in some cases, the first-principles calculation can only represent a trend. In consequence, more optimization is needed in the future in order to achieve results more consistent with actual experiments.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work is supported by the National Nature Science Foundation of China (No. 51778383) and Sichuan Science and Technology Program (No. 2019YFS0500).
| [1] |
Y.Y. Xie, B. Zhao, L. Zhang, R. Luo, Particuology 20 (2015) 141-149. DOI:10.1016/j.partic.2015.01.003 |
| [2] |
S.S. Lim, T. Vos, A.D. Flaxman, et al., Lancet 380 (2012) 2224-2260. DOI:10.1016/S0140-6736(12)61766-8 |
| [3] |
D.Y.H. Pui, S.C. Chen, Z.L. Zuo, Particuology 13 (2014) 1-26. DOI:10.1016/j.partic.2013.11.001 |
| [4] |
G.D. Li, C.L. Fang, S.J. Wang, S.A. Sun, Environ. Sci. Technol. 50 (2016) 11452. DOI:10.1021/acs.est.6b02562 |
| [5] |
J. Tao, K. Ho, L.G. Chen, et al., Particuology 7 (2009) 68-75. DOI:10.1016/j.partic.2008.11.002 |
| [6] |
M. Elser, R.J. Huang, R. Wolf, et al., Atmos. Chem. Phys. 15 (2015) 30127-30174. DOI:10.5194/acpd-15-30127-2015 |
| [7] |
J.J. Cao, Z.X. Shen, J.C. Chow, et al., J. Air Waste Manage. Assoc. 62 (2012) 1214-1226. DOI:10.1080/10962247.2012.701193 |
| [8] |
M N.J., EOS Trans. Am. Geophys. Union 79 (2013) 479-479. |
| [9] |
J. Xue, H. Luo, B.L. Zhang, Adv. Mater. Res. 734- 737 (2013) 1833-1841. |
| [10] |
D. Yu, X.X. Sun, MATEC Web Conf. 64 (2016) 06001. DOI:10.1051/matecconf/20166406001 |
| [11] |
Y.H. Geng, C.H. Peng, M.Z. Tian, Energy Procedia 5 (2011) 370-376. DOI:10.1016/j.egypro.2011.03.063 |
| [12] |
G.R. Parmar, N.N. Rao, Crit. Rev. Environ. Sci. Technol. 39 (2008) 41-78. DOI:10.1080/10643380701413658 |
| [13] |
C.L. Chen, C.H. Wang, H.S. Weng, Chemosphere 56 (2004) 425-431. DOI:10.1016/j.chemosphere.2004.04.006 |
| [14] |
A. Rodas Grapaín, J. Arenas Alatorre, A. Gómez Cortés, G. Díaz, Catal. Today 107 (2005) 168-174. |
| [15] |
J. Xiang, Q.S. Zhao, S. Hu, et al., Asia-Pac. J. Chem. Eng. 2 (2010) 182-189. |
| [16] |
H.X. Jin, L.J. Shi, J.Q. Li, F.Z. Wu, D.W. Tang, Energy Eng. 4 (2003) 33-35. |
| [17] |
Z.L. Ren, X.F. Zhu, W.J. Jiang, D.Z. Dan, Tech. Equip. Environ. Pollut. Control 5 (2004) 90-93. |
| [18] |
Q.Y. Liu, C.H. Li, Y.X. Li, Carbon 41 (2003) 2217-2223. DOI:10.1016/S0008-6223(03)00205-7 |
| [19] |
S. Sumathi, S. Bhatia, K.T. Lee, A.R. Mohamed, J. Hazard. Mater. 176 (2010) 1093-1096. DOI:10.1016/j.jhazmat.2009.11.037 |
| [20] |
A. Lisovskii, R. Semiat, C. Aharoni, Carbon 35 (1997) 1639-1643. DOI:10.1016/S0008-6223(97)00129-2 |
| [21] |
S.J. Allen, E. Ivanova, B. Koumanova, Chem. Eng. J. 152 (2009) 389-395. DOI:10.1016/j.cej.2009.04.063 |
| [22] |
A. Gupta, V. Gaur, N. Verma, Chem. Eng. Process. 43 (2004) 9-22. DOI:10.1016/S0255-2701(02)00213-1 |
| [23] |
J. Tantet, M. Eić, R. Desai, Gas Sep. Purif. 9 (1995) 213-220. DOI:10.1016/0950-4214(95)98229-E |
| [24] |
I. Mochida, Y. Korai, M. Shirahama, et al., Carbon 38 (2000) 227-239. DOI:10.1016/S0008-6223(99)00179-7 |
| [25] |
A. Demirbas, Energy Sources 28 (2006) 1329-1335. DOI:10.1080/009083190910550 |
| [26] |
S.G. Deng, Y.S. Lin, Ind. Eng. Chem. Res. 34 (1995) 4063-4070. DOI:10.1021/ie00038a048 |
| [27] |
O.T. Summerscales, F.G.N. Cloke, P.B. Hitchcock, J.C. Green, N. Hazari, Science 311 (2006) 829-831. DOI:10.1126/science.1121784 |
| [28] |
N.N. Nair, S. Eduard, M. Dominik, J. Am. Chem. Soc. 128 (2006) 13815-13826. DOI:10.1021/ja063295a |
| [29] |
S. Arulmozhiraja, M. Morita, Chem. Res. Toxicol. 17 (2004) 348-356. DOI:10.1021/tx0300380 |
| [30] |
Y. Hong, C. Fairbridge, Energy Fuels 17 (2003) 387-398. DOI:10.1021/ef020171k |
| [31] |
H. Chermette, J. Comput. Chem. 20 (2015) 129-154. |
| [32] |
M.S. Hybertsen, S.G. Louie, Phys. Rev. Lett. 55 (1985) 1418-1421. DOI:10.1103/PhysRevLett.55.1418 |
| [33] |
X.L. Tan, M. Fang, X.K. Wang, Molecules 15 (2010) 8431-8468. DOI:10.3390/molecules15118431 |
| [34] |
V. Gavini, K. Bhattacharya, M. Ortiz, J. Mech. Phys. Solids 55 (2007) 697-718. DOI:10.1016/j.jmps.2007.01.012 |
| [35] |
H.W. Wu, N. Zhang, H.M. Wang, S.G. Hong, Chem. Phys. Lett. 568- 569 (2013) 84-89. |
| [36] |
X.X. Zhang, Z.Q. Dai, Q.C. Chen, J. Tang, Phys. Scr. 89 (2014) 065803. DOI:10.1088/0031-8949/89/6/065803 |
| [37] |
W.K. Heisenberg, Z. Phys. 33 (1925) 879-893. DOI:10.1007/BF01328377 |
| [38] |
W.A. Brown, P. Gardner, D.A. King, Surf. Sci. 330 (1995) 41-47. DOI:10.1016/0039-6028(95)00397-5 |
| [39] |
P.A.M. Dirac, Phys. Today 11 (1958) 32-33. |
| [40] |
A.J. Pounds, J. Chem. Educ. 85 (2008) 919. DOI:10.1021/ed085p919 |
| [41] |
R. Nityananda, P. Hohenberg, W. Kohn, Resonance 22 (2017) 809-811. DOI:10.1007/s12045-017-0529-3 |
| [42] |
L.J. Sham, W. Kohn, Phys. Rev. 145 (1966) 561-567. DOI:10.1103/PhysRev.145.561 |
| [43] |
T. Mineva, S. Krishnamurty, D.R. Salahub, A. Goursot, Int. J. Quantum Chem. 113 (2013) 631-636. DOI:10.1002/qua.24015 |
| [44] |
P. Kaghazchi, J. Phys. Condens. Matter 25 (2013) 095008. DOI:10.1088/0953-8984/25/9/095008 |
| [45] |
A. Débart, A.J. Paterson, J.L. Bao, P.G. Bruce, Angew. Chem. Int. Ed. 47 (2008) 4521-4524. DOI:10.1002/anie.200705648 |
| [46] |
W. Kohn, L.J. Sham, Phys. Rev. 140 (1965) A1133-A1138. DOI:10.1103/PhysRev.140.A1133 |
| [47] |
A.D. Becke, Phys. Rev. A 38 (1988) 3098-3100. DOI:10.1103/PhysRevA.38.3098 |
| [48] |
J.P. Perdew, K. Burke, M. Ernzerhof, Phys. Rev. Lett. 77 (1996) 3865-3868. DOI:10.1103/PhysRevLett.77.3865 |
| [49] |
J.P. Perdew, K. Burke, Y. Wang, Phys. Rev. B 54 (1996) 16533-16539. DOI:10.1103/PhysRevB.54.16533 |
| [50] |
J. Paier, M. Marsman, G. Kresse, J. Chem. Phys. 127 (2007) 024103. DOI:10.1063/1.2747249 |
| [51] |
Q. Xiang, Z.H. Xiong, L.L. Chen, Y. Jiang, Proc. SPIE-Int. Soc. Opt. Eng. 8418 (2012) 127-131. |
| [52] |
J. Hafner, J. Comput. Chem. 29 (2010) 2044-2078. DOI:10.1002/jcc.21057 |
| [53] |
D.E. Jiang, E.A. Carter, J. Phys. Chem. B 108 (2015) 19140-19145. |
| [54] |
L.X. Ling, J.J. Song, S.P. Zhao, R.G. Zhang, B.J. Wang, RSC Adv. 4 (2014) 22411-22418. DOI:10.1039/C4RA02485K |
| [55] |
J.X. Ma, F. Ming, N.T. Lau, J. Catal. 158 (1996) 251-259. DOI:10.1006/jcat.1996.0024 |
| [56] |
J. VandeVondele, J. Hutter, J. Chem. Phys. 127 (2007) 114105-114131. DOI:10.1063/1.2770708 |
| [57] |
J. Hafner, J. Comput. Chem. 29 (2008) 2044-2078. DOI:10.1002/jcc.21057 |
| [58] |
A.V. Slack, H.L. Falkenberry, R.E. Harrington, Air Repair 22 (1972) 159-166. |
| [59] |
J.A. Rodriguez, T. Jirsak, S. Chaturvedi, M. Kuhn, Surf. Sci. 442 (1999) 400-412. DOI:10.1016/S0039-6028(99)00952-8 |
| [60] |
M. Höfer, S. Hillig, H.W. Wassmuth, Vacuum 41 (1990) 102-104. DOI:10.1016/0042-207X(90)90286-8 |
| [61] |
L. Wilde, M. Polcik, J. Haase, et al., Surf. Sci. 405 (1998) 215-227. DOI:10.1016/S0039-6028(98)00050-8 |
| [62] |
K. Wilson, C. Hardacre, C.J. Baddeley, et al., Surf. Sci. 372 (1997) 279-288. DOI:10.1016/S0039-6028(96)01107-7 |
| [63] |
M. Polcik, L. Wilde, J. Haase, et al., Phys. Rev. B 53 (1996) 13720-13724. DOI:10.1103/PhysRevB.53.13720 |
| [64] |
P. Zebisch, M. Weinelt, H.P. Steinrück, Surf. Sci. 295 (1993) 295-305. DOI:10.1016/0039-6028(93)90276-P |
| [65] |
M.L. Burke, R.J. Madix, Surf. Sci. 194 (1988) 223-244. DOI:10.1016/0039-6028(94)91257-2 |
| [66] |
D.A. Outka, R.J. Madix, G.B. Fisher, C.L. Dimaggio, Langmuir 2 (1986) 406-411. DOI:10.1021/la00070a005 |
| [67] |
K. Wang, The Theoretical Study of the Desorption of SO2 and NOx of the Flue Gas on the Copper-based Desulfurizer Dissertation, Taiyuan University of Technology, 2015.
|
| [68] |
G. Pacchioni, Surf. Sci. 281 (1993) 207-219. DOI:10.1016/0039-6028(93)90869-L |
| [69] |
G. Pacchioni, G. Cogliandro, P.S. Bagus, Int. J. Quantum Chem. 42 (1992) 1115-1139. DOI:10.1002/qua.560420504 |
| [70] |
G. Pacchioni, J.M. Ricart, F. Illas, J. Am. Chem. Soc. 116 (1994) 10152-10158. DOI:10.1021/ja00101a038 |
| [71] |
D. Torres, S. González, K.M. Neyman, F. Illas, Chem. Phys. Lett. 422 (2006) 412-416. DOI:10.1016/j.cplett.2006.02.087 |
| [72] |
J.A. Rodriguez, J.M. Ricart, A. Clotet, F. Illas, J. Chem. Phys. 115 (2001) 454-465. DOI:10.1063/1.1377884 |
| [73] |
B. Mortazavi, O. Rahaman, M. Makaremi, et al., Phys. E 87 (2017) 228-232. DOI:10.1016/j.physe.2016.10.047 |
| [74] |
S. Saxena, R.P. Chaudhary, Shukla S., Sci. Rep. 6 (2016) 31073. DOI:10.1038/srep31073 |
| [75] |
X.P. Chen, C.J. Tan, Q. Yang, et al., J. Phys. Chem. C 120 (2016) 13987-13994. DOI:10.1021/acs.jpcc.6b04481 |
| [76] |
X.H. Zha, K. Luo, Q.W. Li, et al., Europhys. Lett. 111 (2015) 26007. DOI:10.1209/0295-5075/111/26007 |
| [77] |
X.F. Yu, J.B. Cheng, Z.B. Liu, et al., RSC Adv. 5 (2015) 30438-30444. DOI:10.1039/C5RA01586C |
| [78] |
M. Khazaei, M. Arai, T. Sasaki, et al., Adv. Funct. Mater. 23 (2013) 2185-2192. DOI:10.1002/adfm.201202502 |
| [79] |
S.H. Ma, D.Y. Yuan, Z.Y. Jiao, T.X. Wang, X.Q. Dai, J. Phys. Chem. C 121 (2017) 24077-24084. DOI:10.1021/acs.jpcc.7b07921 |
| [80] |
S. Bai, L.M. Wang, X.Y. Chen, J.T. Du, Y.J. Xiong, Nano Res. 8 (2015) 175-183. DOI:10.1007/s12274-014-0606-9 |
| [81] |
Y.M. Chen, X.Y. Yu, Z. Li, U. Paik, Lou X.W., Sci. Adv. 2 (2016) e1600021. |
| [82] |
S. Hussain, K. Akbar, D. Vikraman, et al., RSC Adv. 5 (2015) 15374-15378. DOI:10.1039/C4RA14048F |
| [83] |
H.L. Wei, Y.G. Gui, J. Kang, W.B. Wang, C. Tang, Nanomaterials 8 (2018) 646-658. DOI:10.3390/nano8090646 |
| [84] |
J. Zhu, H. Zhang, Y.W. Tong, et al., Appl. Surf. Sci. 419 (2017) 522-530. DOI:10.1016/j.apsusc.2017.04.157 |
| [85] |
K. Czelej, K. Cwieka, K.J. Kurzydlowski, Catal. Commun. 80 (2016) 33-38. DOI:10.1016/j.catcom.2016.03.017 |
| [86] |
Q. Zhang, DFT Study of NH3 and NO Adsorption Behavior on Fe Doping MnO2 (110) Surface Dissertation, Taiyuan University of Technology, 2015.
|
| [87] |
S.L. Liu, Y.W. Li, J.G. Wang, H.J. Jiao, J. Phys. Chem. C 119 (2015) 28377-28388. DOI:10.1021/acs.jpcc.5b07497 |
| [88] |
D.L. Fu, W.Y. Guo, M. Li, H.Y. Zhu, Y.J. Liu, J. Mol. Struct. 1062 (2014) 68-76. |
| [89] |
J.Y. Ye, C.J. Liu, Q.F. Ge, J. Phys. Chem. C 116 (2011) 7817-7825. DOI:10.1021/jp3004773 |
| [90] |
J. Li, DFT Study on the Adsorption and Decomposition of SO2 and NO2 on Metal Surface Dissertation, Southwest University, 2011.
|
| [91] |
D.E. Jiang, E.A. Carter, Surf. Sci. 583 (2005) 60-68. DOI:10.1016/j.susc.2005.03.023 |
| [92] |
L.I. Yia, B. Ding, Chin. J. Struct. Chem. 23 (2004) 1195-1200. |
| [93] |
K.S. Novoselov, A.K. Geim, S.V. Morozov, et al., Science 306 (2004) 666-669. DOI:10.1126/science.1102896 |
| [94] |
Y.N. Tang, Z.Y. Liu, X.Q. Dai, et al., Appl. Surf. Sci. 308 (2014) 402-407. DOI:10.1016/j.apsusc.2014.04.189 |
| [95] |
Y.H. Zhang, Y.B. Chen, K.G. Zhou, et al., Nanotechnology 20 (2009) 185504. DOI:10.1088/0957-4484/20/18/185504 |
| [96] |
H.P. Zhang, X.G. Luo, X.Y. Lin, X. Lu, Y. Leng, Int. J. Hydrogen Energy 38 (2013) 14269-14275. DOI:10.1016/j.ijhydene.2013.07.098 |
| [97] |
J.Y. Dai, J.M. Yuan, Phys. Rev. B 81 (2010) 165414. DOI:10.1103/PhysRevB.81.165414 |
| [98] |
J.Y. Dai, J.M. Yuan, P. Giannozzi, Appl. Phys. Lett. 95 (2009) 232105. DOI:10.1063/1.3272008 |
| [99] |
D. Cortés-Arriagada, N. Villegas-Escobar, D.E. Ortega, Appl. Surf. Sci. 427 (2018) 227-236. DOI:10.1016/j.apsusc.2017.08.216 |
| [100] |
C. Chen, K. Xu, X. Ji, L. Miao, J.J. Jiang, PCCP 16 (2014) 11031-11036. DOI:10.1039/c4cp00702f |
| [101] |
A. Ghosh, K.S. Subrahmanyam, K.S. Krishna, et al., J. Phys. Chem. C 112 (2008) 15704-15707. DOI:10.1021/jp805802w |
| [102] |
O. Leenaerts, B. Partoens, F.M. Peeters, Phys. Rev. B 77 (2008) 125416. DOI:10.1103/PhysRevB.77.125416 |
| [103] |
L. Shao, G.D. Chen, H.G. Ye, et al., Eur. Phys. J. B 86 (2013) 54-58. DOI:10.1140/epjb/e2012-30853-y |
| [104] |
A.S. Rad, Appl. Surf. Sci. 357 (2015) 1217-1224. DOI:10.1016/j.apsusc.2015.09.168 |
| [105] |
H.J. Zhang, W.L. Cen, J. Liu, et al., Appl. Surf. Sci. 324 (2015) 61-67. DOI:10.1016/j.apsusc.2014.10.087 |
| [106] |
Q.X. Zhou, X.Y. Su, W.W. Ju, et al., RSC Adv. 7 (2017) 31457-31465. DOI:10.1039/C7RA04905F |
| [107] |
Y.N. Tang, Z.Y. Liu, Z.G. Shen, et al., Sens. Actuator. B-Chem. 238 (2017) 182-195. DOI:10.1016/j.snb.2016.07.039 |
| [108] |
X.Y. Liang, N. Ding, S. Ng, C.M. Wu, Lawrence, Appl. Surf. Sci. 411 (2017) 11-17. DOI:10.1016/j.apsusc.2017.03.178 |
| [109] |
A.S. Rad, M. Esfahanian, S. Maleki, G. Gharati, Sulfur Rep. 37 (2016) 176-188. DOI:10.1080/17415993.2015.1116536 |
| [110] |
A. Shokuhi Rad, V. Pouralijan Foukolaei, Synth. Met. 210 (2015) 171-178. DOI:10.1016/j.synthmet.2015.09.026 |
| [111] |
X.Y. Liu, J.M. Zhang, K.W. Xu, V. Ji, Appl. Surf. Sci. 313 (2014) 405-410. DOI:10.1016/j.apsusc.2014.05.223 |
| [112] |
S. Iijima, Nature 354 (1991) 56-58. DOI:10.1038/354056a0 |
| [113] |
M. Endo, K. Takeuchi, K. Kobori, et al., Carbon 33 (1995) 873-881. DOI:10.1016/0008-6223(95)00016-7 |
| [114] |
I.M. Sharafeldin, N.K. Allam, New J. Chem. 41 (2017) 14936-14944. DOI:10.1039/C7NJ03109B |
| [115] |
S. Santucci, S. Picozzi, F.D. Gregorio, et al., J. Chem. Phys. 119 (2003) 10904-10910. DOI:10.1063/1.1619948 |
| [116] |
Y. Jiao, Y. Zheng, S.C. Smith, A. Du, Z.H. Zhu, ChemSusChem 7 (2014) 435-441. DOI:10.1002/cssc.201300624 |
| [117] |
Z.Y. Deng, J.M. Zhang, K.W. Xu, Appl. Surf. Sci. 347 (2015) 485-490. DOI:10.1016/j.apsusc.2015.04.116 |
| [118] |
C. Tabtimsai, B. Wanno, V. Ruangpornvisuti, Mater. Chem. Phys. 138 (2013) 709-715. DOI:10.1016/j.matchemphys.2012.12.045 |
| [119] |
A.S. Ghasemi, E. Binaeian, H. Tayebi, Y. Modanlou Jouybari, Int. J. Nano Dimens. 7 (2016) 247-246. |
| [120] |
W. Li, X.M. Lu, G.Q. Li, et al., Appl. Surf. Sci. 364 (2016) 560-566. DOI:10.1016/j.apsusc.2015.12.177 |
| [121] |
M. Yoosefian, M. Zahedi, A. Mola, S. Naserian, Appl. Surf. Sci. 349 (2015) 864-869. DOI:10.1016/j.apsusc.2015.05.088 |