 2019, Vol. 30
2019, Vol. 30 


b College of Chemistry, Beijing University of Chemical Technology, Beijing 100029, China;
c School of Civil and Environmental Engineering, Georgia Institute of Technology, Atlanta, GA 30332, United States;
d College of Engineering and Applied Sciences, and School of Energy Resources, University of Wyoming, Laramie, WY 82071, United States;
e Key Laboratory for Green Chemical Technology of the Ministry of Education, School of Chemical Engineering and Technology, Tianjin University, Tianjin 300072, China;
f Collaborative Innovative Center of Chemical Science and Engineering(Tianjin), Tianjin 300072, China;
g Institute for Superconducting and Electronic Materials, Australian Institute for Innovative Materials, University of Wollongong, Wollongong, NSW 2522, Australia;
h Laboratory of Advanced Materials, Department of Chemistry, Fudan University, Shanghai 200438, China;
i Institute of Fundamental and Frontier Sciences, University of Electronic Science and Technology of China, Chengdu 610054, China;
j Chemical Synthesis and Pollution Control Key Laboratory of Sichuan Province, College of Chemistry and Chemical Engineering, China West Normal University, Nanchong 637002, China;
k Institute of Chemistry, St. Petersburg State University, 7/9 Universitetskaya emb., St. Petersburg 199034, Russian Federation;
l Department of Chemical Engineering, University of Patras, Patras 26500, Greece;
m School of Environmental Science and Engineering, Guangdong Provincial Key Laboratory of Environmental Pollution Control and Remediation Technology, Sun Yat-sen University, Guangzhou 510275, China;
n School of Materials Science and Engineering, Nankai University, Tianjin 300350, China;
o Department of Chemistry, Chinese University of Hong Kong, Hong Kong, China;
p Institute of Nanoscience and Nanotechnology, NCSR "Demokritos", Athens 15341, Greece;
q State Key Laboratory of School of Radiation Medicine and Protection and School for Radiological and interdisciplinary Sciences(RAD-X), Soochow University, Suzhou 215123, China;
r State Key Laboratory of High Performance Ceramics and Superfine Microstructure, Shanghai Institute of Ceramics, Chinese Academy of Sciences, Shanghai 200050, China;
s Center of Materials Science and Optoelectronics Engineering, University of Chinese Academy of Sciences, Beijing 100049, China;
t Key Laboratory of Materials Processing and Mold(Zhengzhou University), Ministry of Education, Zhengzhou University, Zhengzhou 450002, China
Yang Tang, Haomin Jiang, Yongmei Chen*, Pingyu Wan, Maohong Fan*
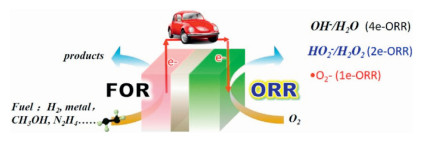
1.1. StatusThe depletion of fossil fuels and pollution of environment have motivated us to develop clean and efficient energy storage and conversion devices. H2-O2 fuel cell, methanol-O2 fuel cell, N2H4-O2 fuel cell and metal-O2 batteries are a series of promising candidates [1, 2]. Electro- catalyzed oxygen reduction reaction (ORR) is the key to those devices. However, the slow kinetics of ORR resulting from the strong bonds of oxygen molecules has greatly hindered the power density and energy efficiency of those ORRrelated electrochemical devices. For example, the overpotential of ORR is as high as 300~500 mV, which is much higher than that (< 50 mV) of hydrogen oxidation reaction in the current H2-O2 fuel cells, making the practical output voltage of single cell 0.6-0.8 V higher than theoretical values. Furthermore, three possible pathways via acceptance of 1, 2 or 4 electrons from cathode for ORR: correspondingly generate superoxide radical (·O2-) and hydrogen peroxide anion/hydrogen peroxide (OOH-/H2O2) as well as hydroxyl anion/water (OH-/H2O), which causes great concern on the selectivity of ORR pathways.
1.2. ORR related applications with different 1e, 2e or 4e pathwaysOwing to the enhanced stability of superoxide radicals (·O2-) in an aprotic ionic liquid (AIL), 1e-ORR could steadily occurs, which is the key cathodic reaction in metal-O2 batteries [3-5]. It was found that 1e-ORR pathway (Fig. 1) in an AIL shifts a 2e or 4e pathway to produce OOH-/H2O2 or OH- when some protic additives [6], water [7-9] or metal ions (such as Mg2+) [10] were present in AIL, or a protic ionic liquid (PIL) was used as the supporting electrolyte [11]. These so called "reactive oxygen species (ROS)", including ·O2-, OOH- or H2O2 formed on site through ORR process, are utilized to electrochemically degrade p-benzyloxyl phenol, a lignin model compound, where ROS attacks C4 atom on the opposite site of the phenolic hydroxyl, resulting in the cleavage of alkyl-O-aryl bond [12].

|
Download:
|
| Fig. 1. Illustration of ORR-related devices. | |
{kind=link}
The 2e-ORR pathway is favorable to the electrochemical production of H2O2 aqueous solution, which is an important chemical as oxidant/reductant in industry, medicine manufacturing, and environmental treatment [13]. As compared with anthraquinone process, electrochemical production of high concentration of H2O2 with high selectivity, activity and longterm durability is still a great challenge.
ORR mainly proceeds via 4e pathway when appropriate catalysts are loaded on cathodes in aqueous solution or polymer electrolytes (proton/alkaline ion exchange membrane). In the case of fuel cell, 4e pathway is an ideal reaction in which 2e pathway is considered as a side-reaction. H2O2 and its generated ROSs can attack the electrocatalyst or carbon substrate or membrane, resulting in the deterioration of these devices.
1.3. Advances in 4e-ORR catalyst and its challengesIn order to decrease the overpotential and improve the 4e pathway selectivity of ORR in fuel cell and metal air batteries, numerous works are focused on preparing high-performance ORR electrocatalyst with high activity, durability and low yield of H2O2. The precious Pt, PtRu, and PtPd alloys are regard as the most efficient 4e-ORR electrocatalysts, but their large-scale applications are greatly hindered due to their high costs. Non-precious metals, such as Co, Ni, Fe or Mo are used to interact with Pt to decrease consumption of Pt. Also, adjusting their compositions will also improve activity and durability. Moreover, the nanostructure engineering of Pt based electrocatalyst in forms of hollow, branch, polyhedron or core-shell can introduce more preferential crystal facet, corner, step or defect, further improve the activity. Huang and the coworkers [14] prepared a series of ternary octahedral MPt3Ni (M = V, Cr, Mg, Fe, Co, Mo, W or Re). Mo-Pt3Ni/C demonstrated the outstanding 4e-ORR performance, with a specific activity of 10.3 mA/cm2 and mass activity of 6.98 A/mg Pt, and improved stability for 8000 cycles. Theoretical calculations suggest that the surface Mo-oxide near the particle vertex/edge sites can be improved from the perspectives of performance and the stability of the Pt3Ni catalyst.
In order to be less dependent on expensive and scarce Pt based materials, nonprecious ones, including spinel oxides, perovskite, MnO2, metal-free heteroatom (N, P, S and B) doped carbon, and nonprecious metal (Fe, Co, Ni and Cu) and heteroatom co-doped carbon (MNC) (Fig. 2), have been extensively investigated [15-18]. Ten years ago, Dai's group [19] reported that metal-free nitrogen doped carbon nanotubes (NCNTs) had lower overpotential, longer operation stability than Pt for ORR in alkaline fuel cells. The relatively high positive charge density on carbon atoms induced by adjacent nitrogen atoms contributes to a 4e pathway for the ORR on NCNTs with excellent performance. Zelenay's group [17] prepared a family of MNC (M = Fe or Co) materials and the highperformance PANI-Fe-C catalyst exhibits a H2O2 yield of less than 1% over the potential range of 0.1-0.8 V (vs. RHE), signaling virtually complete reduction of O2 to H2O via 4e pathway. With respect to MNC electrocatalysts, not only pyridinic-N and graphitic-N, but also MNx and MCx as well as MNxCy have been disclosed as the active sites for ORR by different authors. Recently, in-situ XANES, EXAFS spectroscopy characterization and DFT calculation are used to further understand the ORR mechanism on MNC, which is beneficial to confirmation of the exact active sites in MNC.

|
Download:
|
| Fig. 2. (a) Synthesis summary for typical MNC ORR catalysts. Produced with permission [15]. Copyright 2018, American Chemical Society. (b) Schematic illustration of the formation of the FeN-HPC using NaCl as templates. Produced with permission [16], Copyright 2019, New York, Pergamon Press. (c) Schematic illustration of the synthesis PANI-M-C catalysts. Produced with permission [17]. Copyright 2011, American Association for the Advancement of Science. (d) Schematic illustration of the synthesis of FeISAs/CN. Produced with permission [18]. Copyright 2011, John Wiley and Sons Ltd. | |
{kind=link}
As shown in Fig. 2, pyrolysis of nitrogen-containing organics, transition metal precursors with or without use of template is widely used as a universal method for fabricating MNC. Typical steps include: (1) mixing or coordinating the nitrogen containing organic compounds with transition-metal salt, (2) heat treatment in N2, Ar or NH3 atmosphere, (3) acid washing, and (4) second heat treatment. Numerous MNCs are prepared by this method or the modified methods in the past decade. Metal-organic frameworks (MOFs), which are constituted by organic ligand and metals and characterized with structural diversity, easily functionalized and high specific surface area, have been widely adopted for preparing MNC by pyrolysis recently [20]. By tuning the metal species, organic ligands and pyrolysis procedure, the MOF derived MNC not only possesses uniform active sites and large specific surface area, but also maintains the ordered structure of original MOF. Li's group [18] introduced a MOF derived single-atom FeNC catalyst by using ZIF-8 as molecular-scale cages to trap and separate iron(Ⅲ) in precursor. After pyrolysis, Fe atom was surrounded with four nitrogen atoms to assemble FeN4 coordination site, a key to the excellent ORR performance of the single-atom FeNC with its one half-wave potential (E1/2) being as high as 0.900 V, which outperformed commercial Pt/C and most non-precious-metal catalysts. In the past several years, lots of reported MNCs especially single-atom electrocatalysts have exhibited comparable or even better activities than Pt for ORR in basic media. However, most of them are still inferior to Pt in the acidic electrolyte within the practical pH environment in commercial PEMFC. Currently, larger numbers of researchers are devoted to designing and preparing high performance ORR catalysts in acidic electrolyte with tireless efforts.
1.4. Concluding remarks and prospectsIn summary, the species on active sites determine the intrinsic activity of the electrocatalyst, reflected by the onset potential, half wave potential, tafel slope and H2O2 selectivity. The nano/micro structure of electrocatalyst greatly influences the apparent activity, such as the diffusion limiting current. Those factors will synergistically determine the energy efficiency, power density, long-term durability of the fuel cells. With the tremendous efforts of scientists and the development of in-situ instruments, it is very possible to clearly reveal the complicated mechanism of ORR, design and prepare ORR electrocatalysts with low cost, high activity, long durability for preparation of future fuel cells, metal air batteries and other ORR-related devices.
1.5. AcknowledgmentThe authors greatly appreciate the support from the Beijing Municipal Natural Science Foundation (Nos. 2182046, 2182050).
2. Electrocatalytic oxygen evolution reactionRongrong Zhang, Sana Ullah, Lun Pan, Jijun Zou*
2.1. StatusElectrocatalytic oxygen evolution reaction (OER), an important reaction involved in water splitting and rechargeable metal-air batteries, has been researched since 1830s when Faraday's law of electrolytic equivalent was proposed. The kinetically favorable OER process occurs through multi-step reactions with four electrons transfer as reported by Hoar at 1933. The thermodynamic potential required for OER at 25 ℃ is 1.23 V in acidic solutions (2H2O (l) ↔ 4H+ + O2 (g) + 4e-) and 0.404 V in alkaline solutions (4OH- ↔ 2H2O (l) + O2 (g) + 4e-) [21]. However, the accumulation of energy at each step makes OER kinetics sluggish and results in large overpotential. Therefore, an electrocatalyst is needed to overcome the energy barrier. Generally, a good electrocatalyst should have high quality and quantity of active sites as well as good electronic conductivity, also corrosion resistance and good durability are necessary. Noblemetal-based materials like IrO2 and RuO2 are regarded as most promising for OER in terms of their good activity and stability in both acid and alkaline electrolytes [22]. However, the high price and scarcity are the major bottlenecks in practical applications, which urge developments of strategies and new materials to decrease usage of noble metals and further enhance activities. Transition metal-based (Co, Ni, Mn, Fe, etc.) materials like oxides, (oxy-) hydroxides, sulfides, phosphides, alloys, and carbon-based materials are promising alternative despite the high overpotential and unsatisfied stability. Therefore, recent research mainly focuses on developing catalysts with superior activities by strategies like defect engineering, coordination control, synergy effects, etc. to modulate electron structures of catalysts to reach appropriate adsorption of intermediates. Studies to understand OER mechanisms and real active sites by experiments and theory calculations are also vital for catalysts design. Besides, self-supporting electrodes demonstrate more satisfied activities and stability than powder catalyst electrodes attributed to its 3D structure and high three-phase contact area, which has more potential in industry.
2.2. Current and future challengesCurrently, a great progress has been made in mechanism understanding and catalyst designing. Since 1930s, several OER mechanisms were proposed, from which the metal peroxide path, also named adsorbate evolution mechanism (AEM), has been widely used for metal-based materials. This mechanism involves multiple adsorbed intermediates (*OH, *O, *OOH, *O2) that exhibit highly correlated adsorption strength. Moreover, kinetics of those mechanisms was studied since 1950s by assuming various rate controlling steps (RCS) first and then equilibrium equations were used to calculate the reaction rate, from which tafel slop was found as a constant varying with different RCS. A recently identified mechanism for perovskite oxides, namely the lattice oxygen oxidation mechanism (LOM) involving direct O-O coupling of intermediates and lattice oxygen, shows lower energy barrier for RCS [23]. It is mandatory to understand mechanisms and find out its kinetic pathway on different catalysts. However, the attention of mechanism research on specific catalysts is still insufficient.
Current OER catalysts can be classified into two categories. Noble metal catalysts (e.g., IrO2 and RuO2) are among the most active catalysts reported to date (with overpotentials of ~330 mV and ~270 mV at current density of 10 mA/cm2, respectively). Due to their high cost and low elemental abundance, IrO2 and RuO2- based materials with high surface area-to-mass ratios have been studied. The activities of these oxides depended on particle sizes, crystal structures (rutile or amorphous), surface terminations, and degrees of hydration (IrO2·xH2O or RuO2·xH2O). As reported, rutile IrO2 and RuO2 nanoparticles and thin films with (100) crystal face show better performance than other structures [24]. In addition, some researches focus on using non-noble metal atoms like Co to substitute noble metals to decrease their usage and also to modify their properties further via doping, building complex and so on. Non-noble metal catalysts - transition metal compounds including metal oxides, hydroxides, oxy-hydroxides, sulfides, phosphides, alloys, etc., show promising activities in OER (with overpotentials of ~190-460 mV at current density of 10 mA/cm2) attributed to their adjustable electronic structures [25]. Among these, the perovskite oxides and lanthanide nickel oxides with eg occupancy close to 1.2 and high covalency of transition metal-oxygen bonds demonstrate excellent activities. Besides, coordination unsaturated metal organic frameworks (MOFs) and heteroatom (N, S, P, O) doped carbon materials with intrinsic defects also show considerable activities. The real active sites of most metal compounds are regarded as MOOH (M = Co, Ni, Fe, etc.) in-situ formed under the high potential oxidization process, which is also more active than pre-formed MOOH crystals probably due to crystal defects and multi-substances synergistic effect.
There are some factors influencing the performance of catalysts, such as morphology, crystal phase and facets, eg electronic configuration, strain, magnetism, which are actually electronic structure regulations to make appropriate adsorption energies of each oxygen intermediates to decrease overpotentials. Some regulation strategies like defect engineering, coordination control and multi-atoms synergistic effects are efficient to modulate the electronic structures to enhance conductivity and optimize intermediates adsorption energies. Especially metal vacancy defects can not only induce electron delocalization to increase electronic conductivity but also modulate coordination and electron structure of lattice oxygen to improve H2O activation and optimize adsorption of intermediates [26, 27]. Besides, recent reports show the magnetic enhancement of electrocatalysis by applying moderate magnetic field to anode, on account of electron spin polarization modulation of active sites and intermediate radicals for optimal adsorption [28].
Although great progress has been made in decreasing overpotential of OER by trying a mass of related catalysts, the bottleneck still exists in current catalyst systems for further improving performance. Hence, more open mind should be used to develop new OER pathways beside catalysts design to bypass high energy barrier steps and substantially reduce energy consumption. For example, it will be imaginative to couple OER with analogous mechanism reactions such as Fenton like reactions. Moreover, under conditions with high oxidation potential and alkali solution, many catalysts will transform to real active sites rapidly, but become relatively stable compound after removing the voltage. As a result, the real active sites are still unclear, which is another challenge to understand the mechanism and catalyst design. In addition, stability studies are far from meeting the requirements of industrialization (>1584 h, one quarter) at high current density (500 mA/cm2).
2.3. Advances in science and technology to meet challengesMore high-end instruments and technologies can be used to characterize the structure of catalyst, so that the structurefunction relationship and OER mechanisms can be better understood. Especially in-situ characterizations like XAFS, Raman, FTIR, etc. are good probes to detect reaction intermediates, as well as the in-situ change of catalysts and real active sites. Combined with electrochemical testing techniques, the reaction kinetics of each step can be measured, so that the catalyst can be modified in a targeted manner. DFT is another powerful tool widely used in simulating of material properties, such as conductivity, electron structures, and adsorption/desorption energies.
2.4. Concluding remarks and prospectsOER as an essential but sluggish reaction of hydrogen production and energy storage is widely studied from aspects of mechanism and catalysts design. With the help of in-situ characterizations and theory calculations, structure-function relationship of catalysts and OER mechanisms have been illustrated to some degree, and thereby many promising catalysts have been developed. In terms of application in industry, more attention should be paid to catalysts in acid electrolyte to fit current proton exchange membrane (PEM) in water splitting, and bifunctional catalysts (OER/ORR) in alkaline electrolyte for rechargeable metal-air batteries. Moreover the stability at high current density should be a focus to satisfy requirements of industrialization.
2.5. AcknowledgmentThe authors appreciate the support from the National Natural Science Foundation of China (Nos. 21676193, 51661145026).
3. Electrocatalytic hydrogen evolution reactionMengmeng Lao, Wenping Sun*
3.1. StatusHydrogen generated from electrochemical water splitting, particularly driven by renewable energy resources, will play an important role in the future clean and sustainable energy society. Basically, the water electrolysis process consists of two half elemental reactions: oxygen evolution reaction (OER) and hydrogen evolution reaction (HER). The development of costeffective and efficient electrocatalysts is critical for realizing the large-scale application of water electrolysis for hydrogen production. The electrocatalytic reaction is a heterogeneous process that occurs on the catalyst surface. According to Sabatier principle, an optimized catalyst surface for a given reaction should have a moderate adsorption strength for the reaction intermediates [29]. Volcano plots, where the exchange current densities (i0) obtained from experimental results plotted as a function of the calculated free energy of hydrogen adsorption (ΔGH*), were developed to elucidate the intrinsic activity of various metal catalysts for HER in acidic medium. As shown in Fig. 3a, it is clear that platinum group metals (PGMs) sit at the top of the volcano plot under acidic conditions, demonstrating the highest HER activity [30]. Although descriptors for alkaline HER activity have not been unified, Markovic et al. also built a simple "volcano" to study the alkaline HER activity trend by plotting overpotential as a function of metal-hydrogen binding energy, which also implies that PGMs exhibit the highest activity (Fig. 3b) [31]. The scarcity and high cost of PGM-based catalysts significantly hinders the substantial large-scale application of water electrolysis plants. In order to address this concern, a varieties of catalyst design strategies and catalyst systems have been developed, including but not limited to, single atom-catalysts (SACs) with ultralow metal loading and long-term durability [32], transition metal-based catalysts such as transition metal dichalcogenides (TMDs) [33], and metal-free catalysts such as heteroatom-doped carbon [34]. Although significant research progresses have been achieved, the activity of those new catalyst systems is still inferior to those of PGMs under both acidic and alkaline conditions. Particularly, the development of alkaline HER catalysts is a bigger challenge. The reaction pathways under alkaline conditions are more complicated (Fig. 3c). It is generally proposed that an additional energy barrier for water dissociation needs to be overcome to proceed the subsequent hydrogen production process [35], and it was found that the alkaline HER kinetics could be greatly accelerated by developing hybrid catalysts with the incorporation of a co-catalyst for water dissociation like Ni (OH)2 (Fig. 3d) [36].

|
Download:
|
| Fig. 3. (a) Volcano plot of i0 as a function of ΔGH* for various metal-based catalysts in acidic conditions. Produced with permission [30]. Copyright 2010, American Chemical Society. (b) A volcano plot, measured in 0.1 mol/L KOH (pH 13), of several metals denoting their HER activity, overpotential at 5 mA/cm2 as a function of their calculated M–H binding energy. Produced with permission [31]. Copyright 2013, Serbian Chemical Society. (c) Schematic pathways for hydrogen evolution reaction under acidic and alkaline conditions. Produced with permission [35]. Copyright 2018, Springer Nature. (d) Comparison between activities for the HER, expressed as overpotential required for a 5 mA/cm2 current density in 0.1 mol/L HClO4 and 0.1mol/L KOH for both bare metal surfaces and Ni (OH)2 modified surfaces. Produced with permission [36]. Copyright 2012, John Wiley & Sons, Inc. | |
{kind=link}
3.2. Current and future challenges
Considering the scarcity of PGMs, one challenge is developing efficient catalyst design strategies to substantially reduce PGMs loadings without deteriorating the performance of the catalysts. Secondly, exploiting PGM-free catalysts with comparable activity to that of PGMs is much tougher. For example, the active centers of MoS2 have been verified to be the edge sites. Triggering the activity of basal planes to enhance the catalytic activity is very challenging. Thirdly, it is of great significance to design either PGM- or PGM-free-based heterostructures to address the sluggish kinetics of water dissociation for alkaline HER, and the descriptors for illustrating the alkaline HER activities have not been unified as well. Further, it is also challenging to seek effective approaches that can precisely control and determine the active sites of the catalysts. Moreover, although a large number of electrocatalysts with promising catalytic performances have been reported, the inherent reasons for the enhanced catalytic activity and quantitative structure-property relationships still require indepth understanding.
3.3. Advances in science and technology to meet challengesEngineering PGM-based heterostructures with low PGM loading is no doubt one of the most promising approaches. It has been extensively reported that the unique heterostructures with well-defined interfaces could improve the intrinsic activity, the density of active sites, and performance durability simultaneously. Specifically, designing heterostructures combined decent water adsorption/dissociation capability with favourable hydrogen affinity has been proved to be an appealing way to accelerate the sluggish alkaline HER kinetics [36, 37]. For example, Sun et al. reported a new Pt/Ni(HCO3)2 heterostructure, where Pt nanoparticles (NPs) were uniformly anchored on Ni (HCO3)2 nanoplates, towards efficient alkaline HER [37]. The promoted alkaline HER activity could be attribute to the following reasons: ⅰ) the electron redistribution at the interface of the heterostructure, which optimizes the hydrogen affinity of the Pt surface and thus enhances its intrinsic HER activity; ⅱ) the welldispersed Pt NPs with high exposure of active sites, and the strong interaction between Pt NPs and Ni(HCO3)2 which prevents the aggregation of Pt NPs during long-term operation; ⅲ) the accelerated Volmer step of alkaline HER process due to the strong water adsorption/dissociation capability of Ni (HCO3)2. Furthermore, developing TMD-based heterostructures is supposed to be another profound strategy, where the catalytically inert basal planes of TMDs can be triggered by the electron interaction and synergistic effect, thus promoting the HER activity [38]. The combination of advanced characterization protocols such as scanning transmission electronic microscopy (STEM), X-ray absorption fine structure (XAFS) and density function theory (DFT) computation methodologies could provide detailed information on the atomic coordination and electronic structure modulation at the interface of the heterostructured catalysts, which is of critical importance for acquire deep understanding of the interface chemistry and the underlying structure-property relationships of the catalysts.
3.4. Concluding remarks and prospectsSignificant progress on rational design and development of HER electrocatalysts has been achieved recently. Nevertheless, there still exist many scientific concerns for HER, such as how to construct single/multiple descriptors for elucidating the origin of alkaline HER activity, and how to illustrate the so-called "synergistic effect" regarding the enhanced catalytic activity of heterostructures in a more convincing way. Researchers should pay more attention to taking the advantages of computational chemistry for investigating the energetics and kinetics of this reaction particularly alkaline HER, and to finally establishing reliable principles for guiding alkaline HER catalysts design. The alkaline HER mechanism remains to be comprehensively understood by the development of advanced in-situ characterization techniques along with theoretical calculations. With regard to catalyst design, surface and interface engineering by designing heterostructures would be the forefront. Controlling and understanding the interface chemistry at atomic level of the heterostructures would be vital to unravelling the related performance enhancement mechanism and achieving rational design of efficient electrocatalysts.
3.5. AcknowledgmentWe acknowledge the support from Australian Research Council (ARC) DECRA Grant (No. DE160100596).
4. Electrocatalytic CO2 reduction toward specific productsChao Yang, Gengfeng Zheng*
4.1. statusThe emission of anthropogenic CO2 gas has been increasing, especially in the combustion of fossil fuels. About 880 ± 35 Gt CO2 of the emissions are still in the atmosphere except for those stored on land (plants and soil) and in the ocean [39]. The accumulated CO2 greenhouse gas is important factor that causes the global temperature to rise in recent years. The electrocatalytic CO2 reduction (ECR) reaction has been suggested as a viable alternative that may help to close the anthropogenic carbon cycle and convert electricity to chemical energy in the form of fuels and feedstocks.
4.2. Current and future challengesCompared to the traditional thermo-catalytic transformations of CO2, the ECR uses protons from the water instead of H2 produced by methane reforming in traditional catalysis. Although there are extensive researches focused on the ECR process by experiments and theories, many significant challenges still remain. Electrochemical reduction of CO2 is a multi-step reaction process, leading to complicated products. Among them, CO and formate are the two simplest products of ECR, which only need two proton-electron pairs. These two C1 products possess different critical intermediates, known as *COOH and *HCOO (where * indicates the atom bound to the catalyst), respectively [40]. Multi-carbon products, such as ethylene and ethanol, represent a more attractive feature for ECR due to their higher energy storage capacity and reactive capability. However, the selective formation and subsequent separation of different products, as well as the elucidation of the molecular-level reaction mechanisms place a critical challenge in these processes. As a consequent, the rational design of electrocatalysts toward specific products should be a key principle for high-efficiency catalysts, especially for the multi-carbon products. Moreover, these electrocatalysts generally present a lower product yield, which limits their further application in the industry. Thus, enhancing both the selectivity and activity is the central part of further research.
4.3. Advances in science and technology to meet these challengesCO, as a significant intermediate, is not only evolved by the twoelectron transfer, but also plays an important role in the C-C coupling to form multi-carbon products. For the CO formation, physisorbed CO2 transforms to chemisorbed CO2 (*CO2δ-) by a single electron reaction, subsequent *CO2δ- protonation to form *COOH, and the dissociation of *COOH to form *CO, by the electroncoupled hydrogenation [41]. The first step for the formation of *CO2δ- is always recognized as the rate-determination step (RDS), with a free energy barrier of 0.43 eV (Fig. 4a) [41].

|
Download:
|
| Fig. 4. (a) Reaction pathway of CO2 to C1 products. (b) Edge site weight percentage for a 2 nm wide Au NW and an Au NP as a function of the number of Au atoms. (c) Idealized ratios of edge/corner for the Au NW and Au NP. (b, c) Produced with permission [42]. Copyright 2014, American Chemical Society. (d) Free-energy diagrams for HCOO-, CO and H2 formation on Bi (001) plane. (e) Projected p-orbital density of states (DOS) of the Bi site with OCHO*, COOH*, or H* adsorbates. The Fermi level (EF) was at 0 eV. Ep in OCHO*, and COOH* and H* were highlighted with yellow, blue, and green dashed lines, respectively. (d, e) Produced with permission [43]. Copyright 2018, Nature Publishing Group. (f) Reaction pathway of *CO to C2+ products in different pH scale. Produced with permission [44]. Copyright 2016, American Chemical Society. (g) Schematic illustration of the graphite/carbon nanoparticles/Cu/PTFE electrode. (h) Cross-sectional SEM image of a fabricated graphite/carbon nanoparticles/Cu/PTFE electrode. (g, h) Produced with permission [46]. Copyright 2018, American Association for the Advancement of Science. (i) Selectivity of Cu2O, Ag-Cu2OPS, Ag-Cu2OPB. Produced with permission [47]. Copyright 2017, American Chemical Society. (j) Reaction pathways for ethanol on a Cu (111) surface. Produced with permission [48]. Copyright 2019, American Chemical Society. | |
{kind=link}
Many metal catalysts have been demonstrated to have a good performance for CO2 to CO, such as Au, Ag, Zn, Pd. Au and Ag are noble metal catalysts with high intrinsic selectivity of CO2 to CO. Besides, as these two elements are on the weak bonding side of Cu from the ECR volcano plot, they are easy for *CO desorption to obtain CO product [40]. Based on this, the morphology, grain boundaries and the size effect of Au and Ag have attracted the attention of researchers. For instance, Sun et al. synthesized ultrathin 2 nm Au nanowires supported by carbon, reaching a 94% CO faradaic efficiency (FE) at –0.35 V versus reversible hydrogen electrode (RHE), duo to a high edge-to-corner ratio (Figs. 4b and c) [42]. Considering the expense of the noble metal, carbon- based materials by modulating the electronic properties of adjacent carbons, and single atom materials with tunable coordination environments, have also shown promising CO performances [40]. On the other hand, formate has a high production value per mole electron (16.1×10-3 $/mol electron), with various industrial applications such as a promising chemical for hydrogen storage. In an ECR process, formate is produced through the *HCOO intermediate, bounded to the surface via both oxygen atoms. The P-block metals (e.g., Sn, Bi, In and Pb) are generally the main section for selectivity to formate. For instance, Li and coworkers reported that ultrathin bismuth (Bi) nanosheets prepared by the in-situ topotactic transformation of bismuth oxyiodide nanosheets showed an optimal performance of >90% formate FE over a broad potential (Figs. 4d and e) [43]. Besides, the Pd-based catalyst in ECR is also a research hotspot, which can reduce CO2 to different C1 products, highly dependent on the applied potential, pH, electrolyte concentration, and electrolysis time.
Compared with C1 products, the formation of high-efficiency multi-carbon products are confronting more complicated reaction pathways and more electrons and proton transfer. To date, Cu is the only pure metal that can reduce CO2 to products with >2e transfers. Considering the whole process, the C - C coupling plays a significant role, which involves pathways with *CO intermediates. For instance, Goddard and coworkers proposed a whole pH scale for multi-carbon products by density functional theory (DFT). At an acidic pH, the C2 (or C3) pathways are kinetically blocked. At a neutral pH, *COH (i.e., the protonated *CO) is the common intermediate, followed by a CO - COH pathway to achieve the C-C coupling. At a high pH, the early C - C coupling through adsorbed CO dimerization suppresses the C1 pathways by kinetics, thereby boosting selectivity for multi-carbon products (Fig. 4f) [44]. The effect of the applied potential and *CO coverage have also been studied. At potentials greater than -0.6 V (vs. RHE), CO was dimerized at the lowest activation energy barrier (0.69 eV), and then *OC - CO intermediate is immediately reduced to *OC - COH and *HOC - COH. At this low potential, the hydrogenation steps take place in the ethylene pathway through the Eley–Rideal mechanism with the protons provided by water molecules. With the increasing of potential, the competition of the *H and *CO occurred due to the higher surface protons binding energy. Thus, the mechanism for ethylene production was altered to the *CHO and *CO coupling at potential less than -0.85 V (vs. RHE). At more negative potentials, the much more *H active sites block the CO adsorption, which slows down the *CO dimerization [45].
Based on the perplexing roadmap of the ECR to C2+ products, we must explicitly design our catalysts in more specific product orientation. Ethylene is a strongly demanding precursor for plastics and ethylene glycol production process, which is normally obtained by processes involving repeated cycles of steam cracking at 750 - 950 ℃ followed by a series of quenching, distillation, and recompression. Sargent and coworkers reported that a copper electrocatalyst at an abrupt reaction interface in an alkaline electrolyte can reduce CO2 to ethylene with 70% Faradaic efficiency at -0.55 V (vs. RHE). In such a system with adaptability to abrupt environment, constant ethylene selectivity was achieved for 150 h. Meanwhile, the electrolyzer engineering can help to eliminate the mass transport limitation of CO2 dissolved in the liquid electrolyte (Figs. 4g and h) [46].
Ethanol (C2H5OH) is a fuel with high volumetric energy density nowadays and thus represents a market with high growth potential and profit compared with the common fossil fuels in the transport industry. Lee et al. incorporated Ag into the phaseblended Cu2O-Cu catalyst to get a 34.15% FE for ethanol at –1.2 V vs. RHE. The remarkable effect derived from the geometric characteristics of phase-blended Ag-Cu2O, which inferred that Ag and Cu are closer to each other within an appropriate distance for efficient CO insertion, leads to an enhancement in the product selectivity toward ethanol (Fig. 4i) [47]. Recently, Sargent and coworkers introduced Ag to a Cu catalyst, producing diverse binding sites confirmed by in-situ Raman spectroscopy. Such a Cu-Ag catalyst achieved an FE of 41% for ethanol at –0.67 V (vs. RHE) and provided a new physical picture to design multi-metallic catalysts to control reaction paths in CO2 reductions toward desired products (Fig. 4j) [48].
4.4. Concluding remarksFromwhat have been discussed above, we can realize the ECR is a promising direction for reducing the CO2 level and obtaining many value-added products. Based on different ECR products, extensive research efforts have been invested to design catalysts for different product specificity. In order to bring the technology closer to the industrial level, the key performance index (such as FE, current and stability) of most economically compelling products, such as CO, formate, ethylene and ethanol should be achieved a higher standard. There are several promising strategies to design catalysts oriented to specific products. Combined with the mechanism of a specific product path with the reaction condition, such as applied electric potential, surface coverage, pH, a vast variety of opportunities can be further revealed in this highly promising and important direction.
4.5. AcknowledgmentWethank the National NaturalScience Foundation of China(No. 21773036) for supporting this work.
5. Earth-abundant catalysts for electrochemical N2 reductionQiling Peng, Ting Wang, Yonglan Luo, Xuping Sun*
5.1. StatusNH3 plays a key role in the Earth's ecosystem and is widely used as an activated N2 building block to manufacture fertilizers and other products. Due to its large hydrogen capacity (17.6 wt%) and high energy density (4.3 kWh/h), NH3 is also regarded as an attractive energy carrier to establish a low-carbon society. The ever-increasing NH3 demand has stimulated significant research interest in artificial N2 fixation. Converting N2 to NH3, however, is difficult because N2 is quite unreactive due to its strong N≡N bond (with bond energy of 941kJ/mol), low polarizability, and lack of dipole moment. Currently, industrial-scale NH3 production mainly relies on the Haber–Bosch process operating at high temperature andpressure.This process consumes a large amount of energy with heavy CO2 emission. In this regard, searching an environmentallybenign and sustainable alternative for artifical N2-to-NH3 fixation is of great importance.
Biologically, N2 fixation occurs at ambient conditions enabled by natural nitrogenases in specific bacteria through multiple proton and electron transfer steps with a significant energy input delivered by ATP. Electrochemical N2 reduction has emerged as an environmentally-benignprocess for sustainable NH3 production at ambient conditions, but it is severely challenged by N2 activation and its efficiency strongly depends on the identification of electrocatalysts with high activity for the N2 reduction reaction (NRR) [49, 50].
5.2. Current and future challengesPrecious-metal catalysts perform efficiently to catalyze the NRR, their scarcity and high cost however hinder the widespread uses. Although organometallic catalysts show high selectivity, they not only suffer from limited stability under rigorously reducing conditions, but face difficulty of effective immobilization onto electrode surfaces because of synthesis issues of such complexes. Another big issue lies in the competitive hydrogen evolution reaction which limits the current efficiency for NH3 formation. It still remains a key challenge to design and develop earthabundant heterogeneous NRR electrocatalysts with a high Faradic efficiency (FE) and a large NH3 yield rate for ambient N2-to-NH3 conversion.
5.3. Advances in science and technology to meet challengesMo is not only involved in nitrogenases to catalyze the natural N2 fixation underambient reactionconditions [51], but also proven as the most successful metal for homogeneous N2 functionalization reactions and many Mo-based molecular complexes have been designed for N2 reduction [52]. Although achieving high selectivity, these homogenous catalysts suffer from limited stability under rigorously reducing conditions, and they also face another difficulty in effective grafting onto electrode materials because of synthesis issues of such complexes. These issues have been solved by developing heterogenous nanoelectrocatalysts. Inspired by that Mo and S elements play significant roles in nitrogenases, we first performed theroetical calculations to study the electronic structures of MoS2 and mapped out the energy profile of NRR on MoS2, with a conclusion that the positively charged Mo-edge played the key role to polarize and activate the N2 molecules [53]. To verify the electrocatalytic activity of MoS2, a proof-of-concept experiment was designed by using MoS2 array on carbon cloth as the cathode, although this catalyst only achieves a low FE of 1.17%. Subsequently, we developed defect-rich MoS2 nanoflower through improvement of preparation technology to boost the NRR activity [54]. This catalyst attains a much higher FE of 8.34% with a large NH3 yield of 29.28 mg h–1 mgcat–1 at -0.40 V vs. reversible hydrogen electrode (RHE), outperforming defect-free MoS2 counterpart (2.18% and 13.41 mg h–1 mgcat–1), with strong electrochemical stability. In another study, Zhao and co-workers demonstrated in-operando created strong Li-S interactions empower the S-rich MoS2 nanosheets with superior NRR catalytic activity [55]. These interactions effectively suppress the HER by reducing H* adsorption free energy from 0.03 eV to 0.47 eV, facilitate N2 adsorption by increasing N2 adsorption free energy from -0.32 eV to -0.70 eV and improve the NRR activity by decreasing the activation energy barrier of the reaction control step (*N2 → *N2H) from 0.84 eV to 0.42 eV. Very interestingly, our results also confirm that other Mo compounds are also active to catalyze the electrochemical N2 reduction, including MoO3 [56], Mo2N [57] and Mo2C [58]. Fig. 5 shows the scanning electron microscopy images for these Mobased NRR nanoelectroatalysts.

|
Download:
|
| Fig. 5. Scanning electron microscopy images for Mo-based NRR nanoelectroatalysts: (a) MoS2 nanosheets/carbon cloth. Produced with permission [53]. Copyright 2018, Wiley-Blackwell. (b) defect-rich MoS2 nanoflowers. Produced with permission [54]. Copyright 2018, Wiley-VCH Verlag. (c) S-rich MoS2 nanosheets. Produced with permission [55]. Copyright 2019, Wiley-VCH Verlag. (d) MoO3 nanosheets. Produced with permission [56]. Copyright 2018, Royal Society of Chemistry. (e) Mo2N nanorods. Produced with permission [57]. Copyright 2018, Royal Society of Chemistry. (f) Mo2C nanorods. Produced with permission [58]. Copyright 2019, American Chemical Society. | |
{kind=link}
As one of the cheapest and most abundant metals on the earth, Fe also exists in biological nitrogenases in the form of Fe-protein and MoFe- protein for reduction of N2 to NH3. The Haber–Bosch process uses Fe catalysts to make NH3 from N2 and H2. Fe is also emerged as an interesting metal with catalytic power for electrochemical N2-to-NH3 fixation. Licht et al. reported using Fe2O3 for efficient N2 reduction electrocatalysis in a molten hydroxide electrolyte cell at temperatures ≥200 ℃ [59]. Under ambient conditions, however, carbon nanotube- supported Fe2O3 nanoparticles as a NRR catalyst only achieves a low FE of 0.15% [60]. Fe3O4 is also capable of catalyzing the NRR with a FE of 2.6% [61]. Our recent studies suggests that β-FeOOH nanorod acts as a highperformance NRR catalyst with a FE of 6.7% [62] and its current efficiency for NH3 formation can be further increased to 9.02% by F doping [63]. Besides, Fe3S4 attains a FE of 6.45% for the NRR [64]. V has been found in some organisms such as algae and fungi, and it is directly relevant to the active center or cofactor of nitrogenase. Theoretical [65] and experimental [66] studies suggest that VN is active for electrocatalytic N2 reduction. Both V2O3/C [67] and VO2 hollow microsphere [68] are also proven effectively for the NRR.
Although Mn2+ is not involved in nitrogenases, previous studies suggest that Mn2+ can greatly enhance the catalytic activity of nitrogenases in extracts from the photosynthetic bacterium rhodospirillum rubrum. Although playing an important role in vitro activate nitrogenases for N2 fixation, Mn2+ is not required for the catalysis. In this regard, it is quite interesting to examine the electrochemical NRR behavior of Mn compounds. Our experimental results suggest that MnO particles on Ti mesh behaves as a robust NRR catalyst for high-performance electrohydrogenation of N2 to NH3 with excellent selectivity at ambient conditions, capable of attaining a high FE of 8.02% and a large NH3 yield of 1.11×10–10 mol s–1 cm-2 at –0.39 V vs. RHE [69]. Of note, metal oxides of other metals without any implications for nitrogenases are also able to perform efficiently for N2 reduction electrocatalysis, including Ti [70], Cr [71], Nb [72], W [73], Co [74], Sn [75], etc.
Some progress has also been achieved in metal-free NRR catalysts. We adopted boron carbide (B4C) nanosheets as a catalyst for high-performance electrochemical NRR process at ambient conditions [76]. S has been utilized as an effective dopant to increase the NRR performances of carbon sphere [77] and graphene [78]. Effective N2 reduction electrocatalysis is also enabled by sulfur dots-graphene nanohybrid [79]. B [80] and O [81] are also effective as dopants to boost the NRR performances of graphene. All these carbon nanocatalysts, however, suffer from the involvement of energy-intensive high-temperature thermal annealing processes for material preparation. We also reported a proof-of-concept demonstration of using a chemically oxidized carbon nanotube as a superior NRR electrocatalyst with a high FE of 12.50% and a large NH3 yield of 32.33 mg h-1 mgcat–1 and at -0.4 V vs. RHE [82]. Our recent work further suggests that surface modification of reduced graphene oxide (rGO) by oxygen-rich tannic acid (TA) is also a mild and effective strategy to boost the NRR activity [83]. It is believed that the strong π–π stacking interactions between π-rich TA and rGO brings the TA into very close proximity to rGO, leading to intimate contact of the oxygen groups of TA and rGO, which favors the effective manipulation of the electronic property of rGO. Of note, other non-metal materials like B [84], black P [85], BP [86], BN [87] and carbon nitride [88] are also active for the NRR.
5.4. Concluding remarks and prospectsAlthough tremendous studies have been carried out in electrochemical N2 reduction, this process is still challenged by N2 activation and its efficiency. Such issues can be fixed by introducing heteroatoms or defects to tune the electronic structures, exposing more active sites, and enhancing the adsorption and activation of N2 molecules. Surface/interface engineering would be another effective strategy. Considerable recent attention has focused on screening non-noble-metal catalysts by theoretical calculations, but more detailed experimental evidences still lack. The perfect combination of theoretical and experimental studies is needed to drive continuous progress in this emerging field of electrochemical N2-to-NH3 conversion for applications.
5.5. AcknowledgementThis work was supported by the National Natural Science Foundation of China (No. 21575137).
6. Photoelectrocatalytic O2 reductionAlexander S. Konev, Oleg V. Levin*
6.1. StatusOxygen reduction reaction (ORR) is a key process in fuel cells technology and it also has a strong prospective for waste water treatment through in situ generation of a green and strong oxidant, H2O2. In energy converting systems, such as fuel cells, oxygen reduction reaction occurs at the cathode, either as a four-electron reduction to H2O or as a two-electron reduction to H2O2. Sluggish ORR kinetics fosters the development of cathode materials capable of catalyzing this process. Additionally, ORR may be enhanced by irradiation of the cell with visible light. An impressive number of photocatalytic systems with internal electron transfer has been reported for production of H2O2 via photoinduced ORR (Fig. 6a), with H2O2 concentration approaching 3 mmol/L [89]. Probing of the photoredox reactions in electrochemical cells [90] created a novel concept of photocatalytic fuel cells [91]. Anodic process was proven to account for the observed photoresponse in most of such systems, while cathodic ORR was shown to be independent on illumination [92]. In accord with this, most common architecture of photoelectrochemical cell contains water splitting photoanode, with oxygen reduction occurring as a dark process at catalytic cathode (Fig. 6b). Application of dual-compartment architecture for such cells allowed to raise the H2O2 concentration limit to impressive 61 mmol/L [93].

|
Download:
|
| Fig. 6. Implementation of photoinduced ORR. (a) Photoredox catalysis: O2 is photocatalytically reduced by the catalyst, which is recovered by using a sacrificial electron donor. (b) Photoanodic cell: electrocatalytic reduction of oxygen by dark cathodic process, driven by photoelectrochemical water oxidation at photoanode. (c) photocathodic cell: photoelectrocatalytic oxygen reduction at photocathode, coupled with a sacrificial electron donor oxidation at anode. | |
{kind=link}
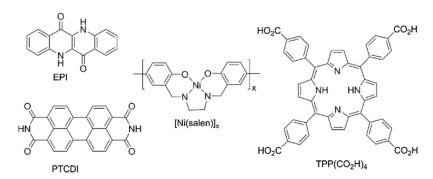
Cells with photoactive cathode (Fig. 6c) are less usual, which is caused by presumed instability of most p-type semiconductors in contact with an electrolyte [91]. To date, true photoelectrocatalytic O2 reduction is reported mostly on organic semiconductors with quinone-like electrochemistry [94], porphyrins [95] and metal salen-type complexes as active redox centers [96] (Fig. 7). The most recent example of organic photoelectrocatalysts reports photocurrent density of ca. 600 μA/cm2 (1 sun illumination, 0.22 V vs. RHE), faradaic efficiency of 60% and H2O2 concentrations of millimolar level [97].

|
Download:
|
| Fig. 7. Examples of compounds used as catalysts for photocathodic ORR: EPIepindolidione, PTCDI- N, N'-dimethyl perylenetetracarboxylic acid diimide, [Ni(salen)]x-polymerized Ni complex with salen ligands, TPP(CO2H)4-5, 15, 20, 25-tetrakis(4-carboxyphenyl)porphyrin. | |
{kind=link}
6.2. Current and future challenges
The efficiency of H2O2 production is governed by the faradaic yield of two-electron ORR vs. four- electron ORR and by the rate of this process measured as photocurrent at photocathode. The reported values of faradaic efficiency in one-compartment cells range typically from 15% to 80%. At least in some cases, the observed non-quantitative faradaic efficiency was shown to be caused by re-oxidation of the produced H2O2 at the anode [98].
Typical photocathodic currents for hydrogen evolution are two orders of magnitude higher than the observed values for H2O2 production (e.g., 22 mA/cm2 vs. 0.1 mA/cm2, 1 sun irradiation, 0 V vs. Ag|AgCl). Taking into consideration concentration and diffusion rate for dissolved O2, theoretical photocathodic current for ORR was predicted by Gryszel et al. to be limited by several mA/cm2, which shows the prospect for the increase of the present values of photocathodic current [94].
In terms of practical applicability, achievement of stable photocurrent response is the important challenge due to the known problem of photocurrent decline during the cell operation [94, 96, 98]. The origin of this problem might be either degradation of the photocathode material [96] or detrimental morphological changes in the photoactive layer [94, 98].
An interesting approach of in situ utilization of superoxide radical-anions generated by photocathodic ORR was suggest by Wu et al., who report on mA/cm2 photocurrent densities for transformation of benzoyl chloride to benzoate by using a PTCD-Ipolythiophentriphenylamine dye as a photoelectrocatalyst of ORR in non-protic medium [97].
6.3. Advances in science and technology to meet challengesPerformance of the photocathodes for H2O2 production was shown to be limited by charge carriers recombination [94]. However, control of the recombination rate by deposition of the electron collecting Au layer on the photoelectroactive material/solution interface, though gave 20-fold increase in photocurrent (to 800 μA), declined the faradaic efficiency to 18%-30%, which is caused by catalyzing of both four-electron and two-electron ORR on Au surface. To favor two-electron ORR, Au layer was covered with organic dyes EPI or PTCDI, that recovered the faradaic efficiency to 60%-80% without loss in photocurrent [94]. Further improvement in faradaic efficiency could be achieved by separating the anodic and cathodic space with a porous membrane to suppress re-oxidation of H2O2 at the anode [98]. Gryszel et al. suggest that utilization of a bulk heterojunction instead of layered architecture should allow for further improvement in the cell performance [94].
A mirror solution to the problem of the simultaneous increase of faradaic efficiency and photocurrent might be implication of dye-sensitized NiO as the photoactive cathode material, where NiO acts as a hole-collecting layer on the pigment/current collector interface and decreases the recombination rate of charge carriers. This allowed to obtain photocurrent of up to 400 μA/cm2 and close to 100% faradaic efficiency in a two-compartment cell [95].
Though significant success has been achieved in affording high faradaic efficiency and photocurrent, securing stability of the photoresponse remains to be mentioned as an issue to be solved, typical reported periods of stable operation of photoelectrosynthetic cells being tens of hours. It should be stressed, however, that the observed decline in cell performance may be caused not only by chemical instability of the material [96], but also by morphological changes of the photoactive layer [94]. Utilization of non-covalent intermolecular interactions like hydrogen bonding can be mentioned as an interesting approach to fix the layer morphology [98], while structural modifications of the photocatalyst to impart hydrophobicity to the material could help to overcome the chemical instability problem [97].
6.4. Concluding remarks and prospectsAt first glance, O2 reduction in photo (electro)chemical cells is a process known for quite a long time. However, most examples involve oxygen reduction as a dark process coupled with photoinduced water oxidation either at anode or at the semiconductor surface. Though true photocathodic oxygen reduction has been known since early 80 s, formulation of the concept of photoelectrocatalytic fuel cells and positioning of H2O2 as a cheap and "green" oxidant catalyzed the recent interest to this topic. To date, organic dyes based photocathode materials showed attractive performance producing H2O2 in millimolar concentrations and giving photocurrent density of up to 800 μA/cm2, which is still below the few milliampere limit based on O2 concentration and diffusion rate. To achieve this goal, suitable photoredox catalysts, including those applicable for solution processing, and favorable architecture of the photoelectrochemical cell are searched. As prospective catalysts, π-extended organics with carbonyl or phenol fragments capable of demonstrating quinone-like redox behavior and showing high light absorption in visible range can be envisioned. Implementation of device architectures developed for photovoltaic devices like bulk heterojunction, p-n junction and dual band gap configurations is expected to constitute future trend.
6.5. AcknowledgmentThis work was supported by the Russian Foundation for Basic Research (No. 18-03-00864).
7. Photoelectrocatalytic H2 productionPanagiotis Lianos*
7.1. StatusPhotoelectrocatalytic hydrogen production involves the use of a photoelectrochemical cell where hydrogen is produced by reduction of protons or reduction of water at the cathode electrode. For this reason, the term "photoelectrolysis" or "photoelectrocatalysis" cell may also apply to the same procedure. In its most common configuration, the photoelectrocatalysis cell comprises two electrodes, one photoanode where oxidation reactions take place and one cathode electrode where reduction reactions take place [91]. In some cases, a photocathode may be used alone or in combination with a photoanode [91, 99]. In the standard configuration, the photoanode carries an n-type photocatalyst, which absorbs photons generating electron-hole pairs. Photogenerated holes are consumed in oxidation reactions while photogenerated electrons move through an external circuit to the cathode electrode where they assist hydrogen production. The efficiency of electron-hole separation depends on many factors, two of them being the most important. Thus the presence of a sacrificial electron donor helps consumption of holes while the existence of sufficient bias drives electrons away and thus prevents recombination. In the case of a photocathode, the photogenerated electrons are consumed in situ to carry out reductions while the holes are transferred through an external circuit to the anode. So far no photocathode has been found which is stable or efficient enough to substitute for the standard photoanode-dark-cathode configuration.
The sacrificial electron donor may derive from a broad choice of organic and inorganic materials. The most celebrated is water itself. Water can be photocatalytically oxidized at the photoanode producing oxygen and be reduced at the cathode producing hydrogen. This challenging project (water splitting) has enjoyed high popularity. However, water oxidation is a 4-electron low probability process thus demanding the presence of oxygen evolution catalysts [91, 99]. On the contrary, oxidation of organic substances is a 2-electron process [91]; therefore, it is easier to oxidize an organic substance than to oxidize water. In addition, oxidation of organics is facilitated in the presence of hydroxyl radicals who may be easily produced by simple one- electron processes OH- + h+ → ·OH. Despite of the tremendous popularity of water splitting, it is more interesting to photocatalytically produce hydrogen by oxidizing an organic sacrificial agent, a process that does notnecessitate anycatalyst other than the photocatalyst itself [100-103]. In addition, the organic substance may be a waste or a pollutant. In that case, a double environmental benefit is gained by degrading a harmful substance and producing renewable hydrogen though photoelectrocatalysis.
7.2. Current and future challengesThe most important factors ensuring cell function is hole scavenging and sufficient bias to drive electrons away from the photocatalyst. If water remains the target, it is necessary to search for oxygen evolution catalysts. However, the matter of hole scavenging is pretty well solved in the presence of organic donors, which offer the above discussed advantages. In this respect, the main challenge remains the complete mapping of the degradation products and the achievement of mineralization [104, 105].
The question of forward bias for electrons is equally important for water splitting as it is for the degradation of organic substances. This is an intrinsic propertyof the cell independentof what the fuel is. In fact no efficient photocatalysts have so far being found which may provide enough potential to drive electrons for hydrogen production. To make it more clear, hydrogen production is obtained at 0 V vs. SHE. The most popular (and most efficient) photocatalysts have a conduction band located either at slightly negative or at 0 V (TiO2 and BiVO4) or even below it at positive potentials (WO3 and Fe2O3). Photogenerated electrons in these photocatalysts cannot drive reduction reactions since they do not have enough electronegativity. Other n-type photocatalysts, like CdS, are better positioned but theyare vulnerable to self-oxidation. This is the reason that researchers have studied p-type semiconductors to make photocathodes. p-type semiconductors do provide electrons with sufficient electronegativity. However, photocathodes alone are no use in organics photodegradation and may only be used in combination with a photoanode. In a standard cell, an external bias is then necessary in order to produce hydrogen. To this end, researchers have examined several approaches. For example, to combine the photoelectrocatalysis cell with a photovoltaic cell or to use tandem cells, one generating the necessary bias and the other producing hydrogen [99]. In this respect, it is attractive to use a photocatalyst that necessitates the lowest possible bias, for example, TiO2 or BiVO4. Anyhow, photoelectrocatalysis is more interesting than electrolysis, which necessitates high voltages and powerful electrocatalysts while photoelectrolysis needs only photocatalysts and small bias ranging below 1 V [91, 99].
Another issue is the efficiency and the light-spectrum span of the photocatalyst. The best photocatalysts in terms of charge carrier mobility are TiO2 and WO3. However, titania absorbs only UV light while WO3 can absorb only a tiny portion of the Visible. This limits their capacity to absorb photons and to produce photocurrent. Visible light absorbing photocatalysts like BiVO4 and Fe2O3 have smaller charge carrier mobility, in particular hematite, and for this reason it must be deposited in very thin films, otherwise it is transformed into an insulator, again becoming weak charge generator. High charge mobility organometallic semiconductors like organometal halide perovskites cannot function in liquid environments, therefore, they are excluded. One solution is to sensitize titania with CdS quantum dots (Fig. 8). We have carried out a systematic study on CdS/TiO2 combined photocatalyst. It functions very well in alkaline electrolytes strictly in the presence of organic fuel. It may also be used in electrolytes containing sulfide salts. It seems then a good choice to spend more effort on improving titania sensitization than waste time on inefficient or exotic photocatalysts. In this respect, I believe that a lot of efforts have been wasted on the search for producing titania in various exotic forms. Nanoparticulate titania can do all the job while titania nanorodes, nanostars or nanoflowers are mere curiosities and offer only a small deal. Visible light absorbing BiVO4 also attracts a lot of interest and for this reason it is very frequently the subject of intensive research [91, 99, 103].

|
Download:
|
| Fig. 8. (a) Light absorption range and (b) photocurrent production by a CdS/TiO2 photoanode in the presence of ethanol in an alkaline electrolyte. | |
{kind=link}
No mention has so far been made to the nature and the materials used for the construction of the (dark) cathode. The cathode electrode must carry an electrocatalyst which facilitates electron transfer from the electrode to the liquid phase. Any electrocatalyst must be characterized by the basic quality triad: large active surface, high conductivity and presence of active sites. These necessary properties are not simultaneously present in untreated materials, hence a great effort is made to develop efficient electrocatalysts. The benchmark choice is, of course, nanostructured noble metals, but they are expensive, limited in quantities and not without problems. This matter concerns not only photoelectrocatalytic hydrogen production but is more important in water electrolysis and fuel cells. Hydrogen production by proton or water reduction is a 2-electron process, therefore, not as difficult to realize as water oxidation. In fact, there are works which describe hydrogen production simply by nanoparticulate carbon electrocatalyst (carbon black) [102]. Such simple electrocatalysts are acceptable because photoelectrocatalysis is a slow process running at small current densities dictated by the light conversion limitations of the photocatalyst. Small current densities result into small hydrogen production rates, therefore, the research on powerful electrocatalysts is welcome but not of prime concern in photoelectrocatalytic hydrogen production.
7.3. Advances in science and technology to meet challengesThe above discussion lets a few issues to emerge as possible targets for the future. Mapping of photocatalytic degradation should be extended to cover all possible organic fuels and conditions for complete mineralization should be ensured. This effort will establish organic substances as first-choice fuel for photoelectrocatalytic hydrogen production. Of course, the ultimate target is the use of wastes or pollutants as fuel. Concerning the choice of photocatalyst, increased effort must be spent on titania sensitization in the Visible while further elaboration on visiblelight absorbing BiVO4 is also realistic. The search for efficient noble-metal-free electrocatalysts will continue, even though, as already said this is not of prime concern to photoelectrocatalytic hydrogen production at the present stage.
It is also necessary to design up-scaled devices. This has already been done in solar cells. In photoelectrocatalysis there exists no experience with up-scaling. Devices remain in laboratory scale so that issues such as charge collection and mass transfer remain at an infant age.
Finally, many researchers recently use photoelectrocatalysis to produce hydrogen peroxide instead of hydrogen, due to the several advantages H2O2 offers as a fuel [106]. In addition, formation of H2O2 does not necessitate a bias since it is formed by oxygen reduction at positive potential thus creating sufficient intrinsic bias. Researchers may then seriously consider this alternative instead of or parallel to hydrogen.
7.4. Concluding remarks and prospectsEnhancement of research on photoelectrocatalysis using organic fuels, search for efficient nanoparticulate titania sensitizers, elaboration on photocatalysts of certified efficiency and upscaling measures together with the study of alternative production of hydrogen peroxide instead of hydrogen may mark the future trends in photoelectrocatalysis.
8. How to improve the efficiency for photoelectrochemical water oxidationZhuofeng Hu*, Zhurui Shen*
8.1. StatusPhotoelectrochemical cells (PECs) are promising technology to convert solar energy into electrical energy or chemical fuel [107]. One important application of PECs is water splitting. Usually, a typical photoelectrochemical cell consists of a semiconductor photoanode, a Pt or graphite cathode, and a reference electrode (applicable). Under illumination, photoexcited electrons and holes are generated in the photoanode. Then, the photoelectrons will migrate to the cathode because the Fermi level of the Pt or graphite is often lower than the conduction band (CB) of the photoanode. As a result, with electrons accumulated, hydrogen could be produced on the cathode. On the photoanode, band bending induces an internal electric field toward the electrolyte. The remaining holes in the photoanode will be driven by the electric field to the interface between the photoanode and the electrolyte. As a result, the hole could oxidize water to produce oxygen. Currently, the most widely-used photoanode materials are metal oxides and sulfide. The photocurrent density is in the range of 1.0 - 8.0 mA/cm2 under AM 1.5 G illumination.
8.2. Current and future challengesThe efficiency of a photoelectrochemical cell is determined by the light absorption efficiency, charge carrier separation efficiency and charge transfer efficiency.
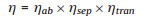
|
(1) |
Where ηab, ηsep and ηsep is the absorption efficiency, separation efficiency and transfer efficiency, respectively.
Nowadays, the promotion of three kinds of efficiencies of PEC above is the main challenge. Absorption is of great significance to the photoelectrochemical cell, which will determine the utilization rate of the solar spectrum. The solar spectrum is mainly composed of ultraviolet light, visible light and infrared light. TiO2, the firstlydiscovered photocatalyst, has been widely used for photoelectrochemical water splitting [108]. It has very high utilization rate of ultraviolet light. However, the ultraviolet light only account for about 5% in the solar spectrum. Besides, charge carrier separation efficiency is another important challenge for the photoelectrochemical cells. When the charges are separated and reach the interface between the photoanode and the electrolyte. Their transfer efficiency determines the efficiency of chemical reaction rate at the interface. For water splitting, the charge transfer efficiency mainly reflects the efficiency of water oxidation rate. However, the kinetic of water oxidation is much slower than that of hydrogen evolution. The water oxidation rate is often considered to be the reaction rate limiting step. It will determine the efficiency of a photoelectrochemical cell.
8.3. Advances in science and technology to meet challenges 8.3.1. Absorption efficiencyIn the past decades, many strategies have been devoted to enhance its solar absorption. Doping is an effective strategy to extend its absorption spectrum [109, 110]. The dopant atoms will cause structure disturbance of the TiO2 and the bandgap of TiO2 become narrow. Also, the dopant will cause dopant level in the middle of the bandgap. These will greatly beneficial to extend the light absorption of TiO2. For example, Horst Kisch and coworkers reports a nitrogen doped TiO2 photoanode with an absorption edge up to 520 nm. This is much wider than pristine TiO2 (320 nm), The efficiency of the photoelectrochemical cell is greatly enhanced [109].
Coupling with other materials with wider absorption spectrum is another effective strategy to enhance the absorption efficiency. Yu and coworkers use a nitrogen-doped carbon layer to extend the photoresponse of TiO2 nanorod array from UV to NIR region. The nitrogen-doped carbon layer could absorb the majority of the solar spectrum. Importantly, the nitrogen-doped carbon layer contact with the TiO2 nanorod array closely, which ensure charge transfer between them. With wider solar spectrum absorption, the photocurrent is superior to that of pristine TiO2 nanorod arrays, and the on-set potential shifted negatively by 0.1 V [111].
8.3.2. Charge carrier separation efficiencyUsually, construction of hetero-structure is a promising method to enhance the separation efficiency, such as CaFe2O4/TaON [112], WO3/BiVO4 [113], Fe2O3/MgFe2O4 [114] and Cr2O3/WO3 [115]. In a hetero-structure, the band diagram of two components of the hetero-structure is different. When they are in contact with each other, internal electric field could be formed and it could promote the charge separation efficiency. Zheng's group construct a WO3/BiVO4 core/shell structure for photoelectrochemical water oxidation. BiVO4 is the primary light-absorber and WO3 acts as an electron conductor. This will dramatically promote the charge separation efficiency. The hetero-structure produces a photocurrent of 3.1 mA/cm2 under simulated sunlight and an incident photon-to-current conversion efficiency of 60% at 300-450 nm, both at a potential of 1.23 V versus RHE [113].
In 2015, Hu and coworkers propose a p-n junction heterostructure to promote charge separation. The Fermi level of p-type semiconductor is close to the valance band, while that of n-type semiconductor is close to the conduction band. When they are in contact with each other, a strong internal electric band could be built, and could remarkably promote charge separation. They build a nanostructured chromium(Ⅲ) oxide/tungsten(Ⅵ) oxide (Cr2O3/WO3) p–n junction photoanode prepared by depositing Cr2O3 nanoparticles onto WO3 nanosheet arrays. Electrochemical and spectroscopic methods indicate that the recombination rate of photogenerated charges becomes lower in this photoanode. Consequently, its onset potential shifts negatively by about 0.1 V and photocurrent density increases from 0.7 mA/cm2 to 1.8 mA/cm2 at 1.8 V vs. RHE. The incident photonto- current efficiency (IPCE) also shows a one-fold improvement. In addition, the construction of the p–n junction leads to an increase of faradaic efficiency (holes to oxygen) from 73.9% to 92.0%, which is attributed to the suppression of side reactions in water oxidation [115].
Besides heterojunction, homojunction is another strategy for charge separation. Homojunction is the junction constructed by the same semiconductors with different band structures [116, 117]. Compared with heterojunction, they are greatly beneficial to charge transfer due to little lattice mismatching and continuity of the chemical bonding. Their morphology, size and defect type determine their efficiency. In 2017, Yu's group diminishes one component of the homojunction to as small as 5 nm by in-situ constructing a QDs- backbone hematite (Fe2O3) homojunction [117]. This novel QDs-based homojunction is formed via intrinsic defects (mainly oxygen vacancies), which is totally different from most reported homojunctions based on extrinsic defects like external doping. With ultrahigh specific surface area, QDs contain sufficient high density of oxygen vacancies to generate "intrinsic" impurity energy levels and form a homojunction. Therefore, the effective hole diffusion length in the QDs-Fe2O3 (0.5–5.0 nm) becomes larger than that of Fe2O3 (0.2–1.0 nm). Consequently, this homojunction shows pronounced enhancement in photoelectrochemical (PEC) performance.
8.3.3. Charge transfer efficiencyDeposition of water oxidation cocatalysts is a generallyaccepted method [118, 119]. Gamelin and coworkers discover a cobalt-phosphate water oxidation catalyst ("Co-Pi") [119]. The cocatalysts can be electrodeposited onto mesostructured Fe2O3 photoanodes. A kinetic bottleneck of water oxidation limiting the activity can be largely overcome by more sparse deposition of Co-Pi onto the Fe2O3. Compared with pure Fe2O3 photoanodes, the sample with Co-Pi loaded exhibit a sustained 5-fold enhancement in the photocurrent density and O2 evolution rate at 1.0 V vs. RHE. It shows that integration of water oxidation cocatalyst with a photon-absorbing substrate can reduce the external power needed to drive the catalyst's electrolysis chemistry.
Besides the kinetic limitation, surface states are another important factor influencing the charge transfer efficiency [120, 121]. The surface of Fe2O3 often contains unwanted surface states. They are mainly attributed to Fe3+/Fe2+ redox couples in oxygen deficient regions (surface oxygen vacancies). They will trap holes or electrons on the surface and cause serious recombination problem during charge/discharge process, thereby inhibiting the increasing of photocurrent density. One method to overcome the surface state is to deposit a passivation overlayers against surface states. Traditional passivation overlayers are metal oxide. However, oxygen vacancies are prevalent for most metal oxides. This is because their formation in metal oxides is often thermodynamically favorable. In contrast, the formation of oxygen vacancies is more energy-consuming when oxygen atoms are covalently bonded. On the basis of this understanding, Shen and coworkers propose a new strategy to transform the surface of Fe2O3 into amorphous iron phosphate (denoted "Fe-Pi"), where the oxygen atoms are "covalently fixed" in phosphate (PO43-). As a result, the oxygen vacancies are decreased and the surface states are effectively suppressed. The onset potential of corresponding photoanode shifts negatively by 0.15 V, and the photocurrent density increases by 4.2 (simulated sunlight) and 4.1 (visible light) times. The suppression of surface states by amorphous Fe-Pi overlayer is then confirmed by series of electrochemical analysis [121].
8.4. Concluding remarks and prospectsOverall, much endeavor has been devoted to develop high efficiency photoelectrochemical cell for water oxidation. Currently, light absorption of photoanode has been extended to near infrared light. Charge separation and transfer efficiency have not yet been reached a satisfactory value. In the future, developing more effective structure and promising materials are the main challenge for photoelectrochemical cells. With the developing of nanoscience and material design, we believe the next generation of photoelectrode could be more and more effective.
8.5. AcknowledgmentThis work is supported by the National Natural Science Foundation of China (Nos. 21872101 and 51902357) and the Start-up Funds for High-Level Talents of Sun Yat-sen University (No. 38000-18841209).
9. Towards artificial photosynthesis: photoelectrocatalytic reduction of carbon dioxideQinglan Zhao, Ying Wang*
9.1. StatusThe increasing accumulation of CO2 concentration in the atmosphere has significant impacts on the global ecosystems, such as global warming and ocean acidification. Searching for renewable and green approaches to utilize CO2 has received significant attention in recent years. One of the most attractive routes is artificial photosynthesis, which is to use sunlight as the energy source to convert CO2 into valuable chemicals and fuels. This also offers an alternative way to store and transport the Sun's intermittent and diffuse energy [122]. The light-assisted CO2 reduction reaction (CO2RR) can be divided into two major categories: in category Ⅰ, the sunlight is directly used with a photosensitizer and a catalyst to facilitate this reaction; in category Ⅱ, solar energy is indirectly utilized by tandeming photovoltaic with electrocatalysis.
For category Ⅰ, the heart is the light driven formation and separation of electrons/holes at the interface. To control the direction of electron flow in the device/assembly is one of the major challenges [123]. In natural photosynthesis, directed electron transfer over long distance is achieved due to the free energy gradient generated from the redox- active group assembly. Back electron transfer is also inhabited because of the exponential dependence of out-sphere electron transfer on distance.
Inspired by the nature, molecular assemblies with more tunable structure and properties are employed to mimic the natural photosynthesis process. Ishitani reported a supramolecular assembly for photoelectrocatalysis of CO2RR, where 80% Faradaic efficiency of CO2 to CO was achieved but the photocurrent was only 8 μA/cm2 [124]. In order to better direct the electron flow, Meyer et al. proposed a chemical approach, "molecular wire", to engineer redox-active group assemblies at a molecular level (Fig. 9) [123, 125]. This analogue contains a well-ordered electron donor (D) and an electron acceptor (A). 85% Faradaic efficiency of CO2 to CO was obtained and the photocurrent for CO2 reduction was improved from ~8 μA/cm2 to 65 μA/cm2 after introducing an electron donor, N, N, N', N'-((CH2)3PO3H2)-4, 4'-dianiline (DA), into the system [125]. Transient adsorption studies suggest the success of directed electron flow in such design. Although there is much progress made in both the mechanism understanding and device building, the high capital cost of these molecules is limiting the further application since a large quantity of the current chromophores and catalysts are based on precious metal, such as Ru, Re and Ir. Researchers in this community started to search for abundant-metal-based alternatives. Reisner et al. reported a high selectivity at 75% of CO on a precious-metal-free catalyst, Co bis (terpyridine), for electrocatalytic CO2 reduction [126]. Ishitani and co-workers developed a series of abundant-metal-based molecules for photocatalytic conversion of CO2 [127]. These efforts advance the progress of replacing precious metal with abundant metal but the remaining work is how to optimize the system to achieve higher efficiency and understanding the mechanism on these molecular analogous.

|
Download:
|
| Fig. 9. Artificial photosynthesis for CO2 conversion. | |
{kind=link}
9.2. Current and future challenges
Heterogeneous material is another major group under the category Ⅰ. Inorganic materials have offered a combined advantage of high efficiency, low cost and long durability [128]. There are several challenges limiting the development of heterogeneous materials. The first one is the photo- adsorption ability, which is how to fine-tune the adsorption region of semiconductors as molecular photosensitizers. The second one, similar to the issue in molecular assemblies, is to control the direction of electron flow in the inorganic materials. This is even more aggravated for heterogeneous materials. The charge recombination happens at an especially short timescale depending on the surface structure. To date, the role of defect on electron- hole separation and recombination is still not clear, with some reports it to be the main factor for the short life time of photogenerated electrons while some holds totally different opinion [129, 130]. Thus, further mechanistic studies to understand interfacial property of semiconductors are needed. Other promising solutions under category Ⅰ is the hybrid systems [131, 132], which need to combine bio-materials, molecular catalysts and inorganic materials. The ultimate goal is to take the advantages from all systems for a higher efficiency. This may provide another route to solve the selectivity and catalytic activity for light-assisted CO2RR.
9.3. Advances in science and technology to meet challengesWe describe category Ⅱ as the "indirect" utilization of solar energy, which is to use solar electricity to facilitate electrocatalysis to reach high conversion of CO2RR. In this configuration, photovoltaic is responsible to harvest the sunlight to electricity [133, 134]. The "solar electricity" is then used to power up an electrolyzer to convert CO2 to other fuels. Indeed, at the current stage, electrocatalysis is much efficient and effective for CO2 conversion [46, 48]. Gratzel et al. reported a tandem system of using a GaInP/GaInAs/Ge photovoltaic with SnO2|CuO as the dark cathode for CO2 reduction to CO in water [133]. A record efficiency of 13.4% solar-to-CO was achieved, which is the one of the best performance for light-assisted CO2RR. This also opens a new route to consider artificial photosynthesis. The ultimate goal of artificial photosynthesis CO2 conversion is to store solar energy in the chemical bond of molecules. The mechanism for electrocatalytic CO2 conversion is relatively well-known as compared to the photoelectrocatalytic system in category Ⅰ. Different products with more than two electron transfer products, such as ethanol and ethylene, were already reported for electrocatalysis. Taking the advantage of the fast development of electrocatalysis, it might be a promising approach to tandem photovoltaic with electrochemical conversion of CO2 for higher energy-containing products.
9.4. Concluding remarks and prospectsProgress has been continuously made to help us understand the role of artificial photosynthesis for CO2RR. How to achieve a high efficiency with a low cost is the major challenge for this system. CO2RR itself is an especially complicated reaction with multielectron and multi-proton transfer. The complexity of the photoinduced electron-holes adds another layer of difficulty for this system. To develop in situ techniques to understand the mechanism of CO2RR and interfacial electron transfer process is rather important in both category Ⅰ and category Ⅱ. System engineering is also another indispensable part of the puzzle. To design a cell for better solar harvesting and overcoming the limitation of mass transport for CO2 molecular in solution is also the key and will need continuous efforts devoting into this field.
10. Photocatalytic hydrogen evolutionNadia Todorova, Christos Trapalis*
10.1. StatusSustainable production of H2 as a clean, carbon-free fuel represents an urgent topic for scientific and technological research. Currently, H2 is mainly produced from natural gas through steam reforming of methane. H2 production using renewable solar energy and abundant raw materials is anticipated to compete the conventional technologies employing non-renewable sources.
After the photo-assisted electrochemical water splitting into H2 and O2, the search for methodologies and catalytic materials operating without application of electrical power and at ambient conditions (pressure, temperature, solar irradiation) is constantly accelerating. By now, water and various organic compounds are studied as sacrificial substances for photocatalytic H2 evolution. In this regard, two main approaches can be distinguished [135]: (ⅰ) photocatalytic pure water splitting giving H2 and O2, and (ⅱ) photocatalytic reforming of organic compounds giving H2, CO2 and H2O. Lately, H2 production from water solutions of sacrificial agents such as methanol, ethanol, amino acids, etc. has been achieved [136, 137]. Increased H2 production has been observed in comparison to the pure water splitting attributed to the Gibbs free energy change and the production of CO2 instead of O2.
Among the different photocatalysts, the TiO2 is the most widely investigated with constant efforts to shift its light absorbance from the UV to the visible part of the solar light spectrum [138]. Also, the CdS has demonstrated remarkable H2 production yield due to favorable position of its band gap edges. In the last years, a new non-metal visible light active photocatalyst g-C3N4 is gathering attention. Recent reports mention significant increase of H2 evolution after its chemical and thermal exfoliation [139]. In most cases, apart from the above-mentioned main photocatalysts, cocatalysts like Pt, Ru/Rh, etc., are employed for effective e--h+ separation [140]. The recent approaches involve adjustment of the band gap edges and controlled charge transfer to increase the H2 yield under solar light. Multi-component photocatalysts containing TiO2, g-C3N4, graphene, CdS, MoS2, WO3, etc., were constructed in various configurations like 2D/2D heterostructures, Z- and S-scheme pairs, etc. [141]. Despite the significant progress made, the efficiency is still far from practical application.
10.2. Current and future challengesA sustainable photocatalytic H2 production process must possess: (ⅰ) low energy consumption, (ⅱ) intensive generation and diffusion of the charge carriers to the surface reactive sites, (ⅲ) formation of appropriate redox species and eco-friendly final products, (ⅳ) stability and repeatability under working conditions, (ⅴ) cost-effectiveness of raw materials and processes. These characteristics are mainly governed by the photocatalyst selected and especially by its electronic properties like band gap width, conductive and valence band edges aligned with the redox potentials of the desired reactions (Fig. 10), as well as its physicochemical and morphological characteristics like affinity, porosity, etc. Hence, the current and future challenges are mainly related to the material engineering and manufacture of visible light working photocatalysts. Although the absorption of TiO2 has been shifted to the larger wavelengths, satisfactory visible light activity remains a challenge. The highly effective for H2 production CdS is prone to photo-corrosion and thus regarded as non-acceptable for environmental reasons. The yield of more recently suggested photocatalyst g-C3N4 is also a challenge since expensive cocatalyst (Pt) and sacrificial agents are still required. The high cost of the photocatalysts and the low density of active sites on its surface have also to be encountered by finding non- expensive and nonhazardous porous supports.

|
Download:
|
| Fig. 10. The mechanism of photocatalytic H2 production. Produced with permission [140]. Copyright 2019, Cell Press. | |
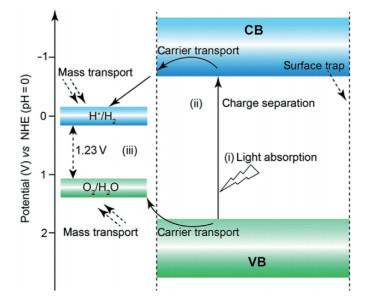{kind=link}
The H2 source also influences the sustainability of the H2 production process. Currently, pure water and organic pollutants are the most attractive H2 sources because clean products and water purification are gained along with the H2 production. However, the highly demanding 2e- oxidation half-reaction constitutes a bottleneck for pure water splitting since there are few narrow band gap photocatalysts that can catalyze both (proton reduction and water oxidation) half-reactions. The utilization of water dissolved organic pollutants as H2 source is considered thermodynamically less demanding than pure water splitting. However, finding abundant and continuously available sacrificial agents, giving low CO2 evolution, produced without competing land for food production, and of low cost, remains a challenge to be faced.
10.3. Advances in science and technology to meet challengesVarious strategies have been employed to improve the light absorption efficiency and charge separation of the photocatalysts. As the narrowing of a semiconductor's band gap is accompanied with decrease or even loss of redox ability, multi-component systems consisted of compatible semiconductors and below-3D materials (quantum dots, nanoparticles, 2D conductive materials) are being constructed in the form of various (Z-, S-) schemes and heterojunctions. The lifetime of the excited cites and the charge carriers transfer were increased by surface facet engineering, loading with co-catalyst, creating heterostructures, etc. Thus, a rate of H2 evolution up to 253.5 μmol g-1 h-1 from pure water under solar light was recorded over Au/TiO2 nanostructures owing to controlled spatial charge separation and redox reactions [142]. The coupling of conventional photocatalysts such as TiO2 and g-C3N4 with carbon nanostructutes (CNTs, graphene, reduced graphene oxide) and other 2D materials increased significantly both visible light absorption and charge separation. The close contact between the components was found to play major role for the efficient transfer of the e- and h+ to the active sites thus preventing their recombination. Also, great attention has been given to the g-C3N4 and especially its exfoliated form, as a narrow band gap, low-cost photocatalyst. Remarkable H2 evolution rates of 830.1 and 115.5 μmol g-1 h-1 under solar and visible light irradiation, respectively, have been reported owing to specific textural and electronic features like high surface area, ultra large pore volume, conductivity, etc. [143]. Defects' engineering, creation of carbon vacancies, modification of band edge potentials and increase of the active sites on the g-C3N4 nanosheets are believed to further improve the yield under solar light irradiation.
Regarding the simultaneous photocatalytic H2 production and water purification from pollutants like alcohols, organic acids, etc., it was found that the H2 evolution mainly depends on the nature of the sacrificial agent, its concentration and the co-catalyst used. Higher H2 evolution has been observed from waste compounds with simple molecule structure that contains hydroxyl groups. Apart from noble metals (Pt, Ag, Au, Ru, etc.) with sub-nano dimensions, transition-metal oxides, hydroxides and sulfides were reported to reduce the activation energy of the H2 evolution. Recent theoretical studies revealed that MXenes, a new family of 2D materials, are promising candidates for catalyzing H2 evolution reactions. Latest experimental results on Ti3C2Tx MXene used as co-catalyst (3 wt%) in presence of sacrificial agent demonstrate significant H2 yield of 6.979 mmol h-1 g-1 as evident in Fig. 11 [144].

|
Download:
|
| Fig. 11. Photocatalytic H2 production activity (a), H2 production rate of TiO2, Ti3C2 and TiO2/Ti3C2 composites (b), recycling photocatalytic H2 production experiments of TT3 sample (c). Produced with permission [144]. Copyright 2019, Elsevier. | |
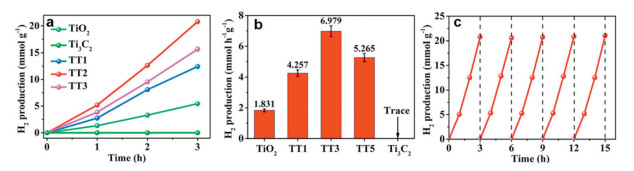{kind=link}
10.4. Concluding remarks and prospects
The photocatalytic H2 production is addressing two major issues of the modern society, i.e., constantly increasing energy demand and environmental pollution. The H2 itself is a clean fuel the products of which do not include hazardous organic substances or CO2 and therefore it is considered an alternative to currently used hydrocarbons like natural gas, petroleum, coal. The use of H2 as a fuel will drastically reduce the damage on the environment caused from the exploitation of conventional energy sources. This effect would be realistic, if the methods for H2 production are clean and environmentally friendly as well. The photocatalytic production of H2 using renewable energy and clean precursors like solar light and pure water represents a sustainable procedure that is being even further optimized by using organic pollutants as H2 source. The process is ruled by the photocatalytic materials and systems that utilize the solar energy to initiate the targeted chemical reactions. Therefore, efforts are concentrated on development of highly effective, stable, non-toxic and low-cost photocatalysts operating at ambient conditions. The most studied are modified TiO2, g-C3N4, below-3D carbon nanostructures and heterostructures that appeared the most promising as well. Novel 2D MXenes are explored as efficient components in composite photoactive systems.
From the above, it is clear that the prospects for the photocatalytic H2 production are towards finding effective ways for charge separation and e- transportation to H2 evolution sites before the process becomes sustainable and industrially applicable.
10.5. AcknowledgmentThe authors appreciate the financial support from "2D NanoSmart" project funded by Stavros Niarhos Foundation and NanoPhos S.A.
11. Photoreduction of dinitrogen to ammoniaMatthew V. Sheridan*
11.1. StatusThe goal of photochemical (PC-) and photoelectrochemical (PEC-) nitrogen reduction reaction (NRR) to produce ammonia is driven by the need to replace the Haber-Bosch process currently responsible for worldwide nitrogen fixation. This industrial process consumes roughly 1%-3% of the world's energy and 3%- 5% of natural gas used each year, and contributes to roughly 2% of the total yearly CO2 released into the atmosphere. Although nitrogen fixation is thermodynamically negative (N2 + 3H2 → 2NH3, ΔH = -92.4 kJ), high temperature and pressures are needed to drive the equilibrium favorably towards ammonia, resulting in a high energy cost for this process of ca.10 eV per mol NH3 [145]. The production of ammonia serves a vital function providing nitrogen fertilizer for agriculture worldwide. The possibility of using sunlight to develop a green technology to drive NRR, and replace the expensive operation, transportation, and infrastructure investment associated with the Haber-Bosch process, is one of the central challenges in chemistry for the 21st century.
Solar fuels and photosynthesis devices have blossomed over the last decade as a way of taking advantage of clean energy from the sun. Much of this work has focused on light-driven water splitting (2H2O → O2 + 2H2), with fewer research works focused on lightdriven organic transformations, and, even rarer, are examples that perform transformations with CO2 and N2 as a chemical feedstock. These latter transformations are obfuscated by product selectivity and, more importantly, the competing proton reduction reaction. A significant challenge in NRR has been finding catalytic surfaces with favorable interactions with dinitrogen to weaken the N, N triple bond from (945 kJ/mol), and function as effective NRRcatalysts. Traditionally, catalysts composed of Fe, Ru, Mo and Pt metals have been developed for NRR. More recently, bismuth materials, such as bismuth oxyhalides, have emerged as promising candidates to activate dinitrogen [146]. These materials promote elongation of the N, N triple bond from 1.078 Å in N2(g) to 1.133 Å for the adsorbed N2 at an oxygen vacancy (OV) on the BiOBr {001} surface. Under visible light illumination, this engineered surface has a high rate of PC-NRR of 104.2 mmol h-1 g-1 (Fig. 12) [147].

|
Download:
|
| Fig. 12. (a) Activation of N2 at an OV in BiOBr {001}; (b) The lower thermal barrier to NRR at the OV; (c) Solar water splitting and nitrogen fixation based on semiconductor photocatalysis. Produced with permission [146]. Copyright 2017, American Chemical Society. | |
{kind=link}
11.2. Current and future challenges
Light-driven ammonia production has been extensively studied for PC-NRR systems in solution. The desired photoreaction, N2 + 3H2O → 2NH3 + 3/2O2, requires a photocatalyst which can undergo photoexcitation events to produce electron-hole pairs that have sufficient energy to catalyze NRR (N2 + 6H+ + 6e- → 2NH3; E = -0.17 V) and water oxidation (2H2O → O2 + 4H+ + 4e-; E = 1.23 V). Fortunately, the moderate energy difference (ΔV) between the two reactions (ignoring overpotential) in aqueous solution (ΔV = 1.4 V) and in the gas phase (ΔV = 1.17 V) is readily accomplished by many semiconductors. The first photocatalyst reported to perform PC-NRR in 1977 was the prototypical semiconductor TiO2, which has an appropriate band alignment and bandgap energy of ca. 3.1 V to perform NRR and water oxidation [148]. Since then, TiO2-based photosystems have continued to be the most well-studied along with, arguably, bismuth systems and graphitic carbon nitride (g-C3N4). Numerous other metal oxide, metal sulfides and chalcogels, carbonaceous material, and ternary semiconductors have also been reported to accomplish PC-NRR [149].
To avoid increasing the N2 pressure (an effective way to boost activity), efforts to increase the chemisorption of N2 to develop better photocatalysts has been the subject of a great number of studies. Efforts to increase adsorption by structural engineering (crystal phase, facet, and vacancies), co-catalysts and dopants, and different morphologies to enhance the surface area have all been pursued as a means to favor greater interactions with N2. To promote longer charge separation, a necessary consequence of the NRR's multi-electron chemistry and slow catalysis, heterojunctions or Z-schemes involving multi-photocatalytic systems have been employed [150]. Plasmon nanoparticles, a common tool in photochemistry, have also been investigated and show enhanced ammonia production on TiO2 photocataysts [151, 152]. Many of the current PC-NRR systems take advantage of one or several of these design principles (increased chemisorption, increased charge separation, and plasmon effects) to achieve more substantial ammonia production.
In PEC-NRR, the NRR and oxidation reactions are spatially separated at two electrodes, with one electrode the NRRphotocathode. This arrangement is preferable to avoid redoxelimination reactions between oxidized and reduced species that lead to a decrease efficiency; a problem commonly witnessed in PC-NRR, where both reactions occur in close proximity. A promising result in this field has been accomplished using a silicon-type photocathode and gold plasmon nanoparticles together with a Cr co-catalyst, a PEC-NRR rate of 13.3 mg m-2 h-1 was obtained under 2 suns illumination [153]. Only a handful of reports on PEC-NRR have been reported for PEC-NRR to date, and the observed photocurrents are still quite low, near or less than 10 μA/cm2.
An essential component in P(E)C-NRR is the catalyst that can be integrated into a photochemical system. A diverse number of materials have been screened in NRR-electrocatalysis, and many have low overpotentials (< 0.4 V), but nearly all suffer from Faradaic efficiencies, FE, of less than 10%, and low catalytic activity (current densities ca. 1-10 μA/cm2) [154]. In some cases, ammonia production rates are greater (ca. 1 mA/cm2) with higher overpotentials, but low to moderate FE still persist [155, 156]. A promising approach to improve FE and current density is to promote selectivity for N2 reduction over proton reduction by reducing the accessibility of protons at the catalyst surface. This can be accomplished by careful solution engineering using non-aqueous electrolytes and organic proton donors [156]. In PC-NRR at Bi nanocrystals, using competing ion equilibria to affect the solution, a high concentration of potassium ion increases FE to 66% along with increasing ammonia production to the relatively high rate of 200 mmol g-1 h-1 [157]. Other methods to modify proton access at the surface include using ionic liquids (ILs), and NRR-electrocatalysis of FE of ca. 60% has been achieved [158], or using polymer membranes that reduce the proton gradient near the surface; a PEC-NRR cells using a poly (tetrafluoroethylene) membrane has achieved a FE of ca. 38% [159].
11.3. Advances in science and technology to meet the challengesThe success of P(E)C-NRR is closely tied to advances in NRRelectrocatalysts. The development of more effective NRR-catalysts will have a significant impact going forward. With increasing maturity, many of the advances being found in water splitting and CO2 reduction are expected to carry over to P(E)C-NRR; for example: advances in charge separation, light harvesting, tandem solar cells, and molecular complexes that can assist in steps along the photochemical pathway. Integration of molecules and semiconductors is routinely used in dye-sensitized solar cells (DSC) and dye-sensitized photoelectrosynthesis cells (DSPEC) for water splitting and CO2 reduction; taking advantage of the large number of molecules capable of binding and activating N2 and, even, complexes that can perform NRR- catalysis, is therefore expected to contribute to greater P(E)C-NRR performances in the future [160].
Low quantum efficiencies observed in PC-NRR can be improved by exploring a number of state-of-the-art heterojunctions for longer charge separation lifetimes. This is one method that can directly make up for slower rates of NRR catalysis. The use of electron-transfer mediators in the form of mixed metal oxides or molecules with different band/HOMO-LUMO energies level and conductors like graphene, which has already found use in PC-NRR to facilitate charge separation, are viable platforms for improving NRR systems via longer charge separation [161]. Further work to understand the fundamental chemistry involved in P(E)C-NRR by experimental and computational chemistry will also aid the progress, such has been the case in light-driven water splitting [162]. Technologies that would increase N2 interaction, by increasing the surface area of the catalyst, are particularly attractive, and metal organic frameworks and coordination network polymers are popular potential candidates [163], along with single atom catalysts [164]. Enhanced activity with greater surface area was demonstrated at bismuth oxide quantum dots whose ammonia production rate (1226 μmol g-1 h-1) was nearly 1000 times higher than in optimized Fe-doped TiO2 photosystems [165]. Lastly, re-exploring f-element photochemistry, where a number of useful photocatalysts with high reduction potentials and long excited-state lifetimes exist, will likely find a place in this field as previous work using Sm oxides with PC-NRR were found to be more effective than iron-based photocatalysts popular at the time [166].
11.4. Concluding remarksThe promise of successful P(E)C-NRR is a scalable nitrogen fixation technology with a low energy input (sunlight) and lower initial investment. For cost competitiveness, the devices should have >1% solar-to-ammonia efficiencies, take advantage of earth abundant minerals, and, preferably, have 10 year lifetimes to match many of the standards already established in the solar field. Significant progress has to be made in many facets of P(E)C-NRR with higher activity and more selective catalysts greatly holding back current photosystems. Finally, due to the often low production yields of ammonia, the community must continue to make particular care to ensure detected ammonia is the result of NRR and not contaminants. Isotopic labelling (15N) is considered the most rigorous experimental evidence for ammonia levels below 1 ppm which would include most results to date [167].
12. Photocatalytic CO2 reduction: Challenges and prospectsHaipeng Wang, Ling Zhang, Songmei Sun, Wenzhong Wang*
12.1. StatusThe concentration of carbon dioxide (CO2) in the atmosphere already exceeded 400 ppm in 2015 and will grow continuously in recent decades. In 2017, global CO2 emissions from fossil fuels combustion reached 33 gigatons, which is twice the natural rate at which CO2 is absorbed back into land and ocean sinks. Reducing the CO2 level and achieving the balance of carbon cycle are the common goals of the world. Photocatalytic CO2 reduction has been regarded as one of the most promising strategies to address environmental and energy issues. Using sunlight as the energy source, CO2 and water are converted into "solar fuels" and oxygen, which not only realizes energy supply but also reduces CO2 concentration. Recently, liquid sunshine was proposed to describe the produced green liquid fuels by photocatalysis, and its future application potential and value are depicted in detail [122]. In the past 40 years, to improve the performance of CO2 reduction, extensive efforts have been devoted to explore the photocatalysts and reaction mechanism. Various catalysts, like metal oxide, metal sulfides, metal nitride, Bi-based material, organic polymer, etc., have been developed for CO2 photoreduction. Although some progress has been made, there is still a long way to go before realizing large scale liquid sunshine production. Therefore, we outline challenges and potential advances associated with photocatalytic CO2 reduction for future commercial applications.
12.2. Current and future challengesThere are four main challenges for the photocatalytic CO2 reduction.
12.2.1. How to enhance light harvesting and charge- separationEfficient light harvesting ensures the generation of abundant photogenerated carriers, which is the basis of photocatalytic reaction. However, at present, the photocatalysts with suitable band structure that not only meet the thermodynamics of CO2 reduction, but also enhance the light absorption range to longer wavelengths, are rare. TiO2, one of the most widely studied materials with a bandgap 3.2 eV, only absorb ultraviolet light which accounts for 4% of the total solar energy. What is more, only when photogenerated carriers are separated and migrate to the surface of the catalyst can the redox reaction occur. However, the charge recombination process (~10-9 s) is much faster than the surface redox processes (10-8-10-1 s), resulting in the loss of photogenerated carriers and a decrease of catalytic efficiency [168].
12.2.2. How to improve the efficiency of CO2 photoreductionPhotocatalytic CO2 reduction involves two half reactions, namely CO2 conversion and water oxidation [169]. Generally, 1.23 V is required to split water thermodynamically, with a similar potential needed for CO2 reduction. However, due to kinetic constraints, potential needed is greatly increased, especially for oxidative half reactions. It is recognized that water oxidation is the rate-limiting step in overall reaction, and there is no effective method to accelerate this step. Furthermore, CO2 is a highly stable linear molecule that is hardly activated and is difficult to be dissociated due to its high C = O bond energy (750 kJ/mol), making the reduction of CO2 thermodynamically and kinetically unfavorable. Therefore, enhancing the oxidation of water and the adsorption and activation of CO2 are the key to improve the photocatalytic efficiency.
12.2.3. How to enhance the selectivity of products, especially selective reduction of CO2 to multicarbon productsFirstly, the HER usually competes with CO2 reduction due to their close reduction potential. Besides, the products of CO2 reduction are very diverse because of the multi-electron reaction process, CO, HCOOH, CH4, HCHO, CH3OH etc., may be generated at the same time. Unfortunately, these products, with little practical value in the mixed state, are difficult to separate. What is more promising is the direct reduction of CO2 to multicarbon oxygenates and hydrocarbons, but it still faces the problem of poor selectivity and low yield. Enhancing the understanding of reaction pathways and the adsorption modes of reactants and intermediate species may help to design highly selective catalysts [170].
12.2.4. How to utilize low concentration CO2 directly.Currently, high purity CO2 is often used as gas source in photocatalytic CO2 reaction. However, the CO2 concentration in the atmosphere is relatively low, about 400 ppm, and even in exhaust gases discharged from industrial furnace, the concentration is about 10%–15%. Purification and concentration of low concentration CO2 often consumes a lot of energy, resulting in new carbon emissions. High-efficiency and high-selectivity CO2 photocatalytic reduction under low CO2 concentration is of great significance for the comprehensive control of CO2 emission, and also remains a challenge [171].
12.3. Advances in science and technology to meet challengesIn recent years, progresses have been made in the development of novel photocatalyst systems and catalytic mechanism.
12.3.1. Surface modificationVarious cocatalysts have been developed to modify the photocatalysts [172]. Metal-based cocatalysts, especially organic metal complex cocatalysts like porphyrins and phthalocyanine, exhibit enhanced preformation in enhancing solar-light absorption. Metal-free cocatalysts, such as CNT and graphene, show their unique advantages, like boosting charge separation and enhancing the stability of photocatalysts, etc. Biological cocatalysts, such as enzyme and bacteria, coupled with inorganic semiconductor photocatalysts, have been proven to be highly active and selective for facilitating photocatalytic CO2 reduction.
12.3.2. Single-atom catalysis (SACs)SACs provide isolated mononuclear metal active sites and the exact same reaction microenvironment for every reactant molecule, which means the intermediate species and products produced by each reaction site are the same. The unique structure of SACs provides a platform to study the reaction pathways and constituent- activity relations accurately. SACs with almost 100% atom utilization efficiency have shown promising application prospects in photocatalytic CO2 reduction [173].
12.3.3. Defects engineeringIntroducing defects in the bulk or surface of the catalyst, including anion and cationic vacancy, atomic doping, etc., enhance the light absorption, promote carrier separation, and especially provide high activity reaction sites. The oxygen vacancies on the surface of TiO2, BiOX (X = F, Cl, Br or I), WO3, etc., have been widely studied, and is effective in promoting the performance of photocatalytic CO2 reduction. Moreover, the carbon vacancy, nitrogen vacancy, sulfur vacancy, metal ion vacancy, etc., have also been developed.
12.3.4. Low-dimensional materialsOwing to distinctive electronic structures and high specific surface areas, low-dimensional materials have recently emerged as a class of potentially inexpensive catalysts with enhanced activity and improved selectivity. 2D materials have attracted the most attention for the better stability and excellent photoelectric properties [174]. Firstly, the optimal behaviors of charge carrier could be predictable, like the reduced migration distance from bulk to surface and the high conductivity. Furthermore, as for the ultrathin feature of 2D materials, the rich reaction site resulted from the high specific surface area would also facilitate photocatalytic reactions.
12.3.5. Photoelectric synergistic catalysisIn the photo-electrocatalytic system, a small applied bias voltage could effectively reduce the recombination of photogenerated carriers and promote the separation of electron and holes. Besides, due to the cathode and anode are isolated in the reaction system, the products at the oxidation side and the reduction side are separated, then the back reaction is also suppressed.
12.3.6. The development of operando characterization techniquesCurrently, some operando techniques (e.g., in situ FTIR, in situ TEM, in situ XANES, in situ EXAFS, in situ XPS, and in situ ESR) have been developed to investigate the reaction pathways, intermediate species, change of catalyst structure and to determine the real active sites [175]. It is of great importance to collect the real information about the catalytic reaction process, which would provide guidance for the design of high- performance CO2 reduction catalysts.
12.4. Concluding remarks and prospectsThe challenges and progress in recent years about photocatalytic CO2 reduction have been outlined. The development of catalyst systems and various modification methods have improved the performance of photocatalytic CO2 reduction. Advances in characterization techniques also provide more accurate information for analysis of reaction mechanisms. Although considerable progress has been made, many major scientific and technical challenges still need to be overcome in order that liquid sunshine is applied widely in our future society. Some prospects are as follows:
Firstly, based on theoretical calculations, large-scale screening of catalysts that meet the requirement of photocatalytic CO2 reduction should be achieved, meanwhile, theory and models should also be improved [176]. Secondly, operando characterization techniques should fully simulate the experimental conditions with simultaneous regulation of atmosphere, temperature, light source, pressure, and reaction medium. Thirdly, the establishment of internationally unified experimental standards - Light sources, reactors, test procedures, and performance evaluations should all have specific standards so that the reaction rates reported by different groups can be directly compared.
12.5. AcknowledgmentThe financial support from the National Natural Science Foundation of China (Nos. 51772312, 21671197) is acknowledged. Declaration of competing interest There is no competing interest for this manuscript.
| [1] |
Z. Wang, J. Parrondo, C. He, S. Sankarasubramanian, V. Ramani, Nat. Energy 4 (2019) 281-289. DOI:10.1038/s41560-019-0330-5 |
| [2] |
M.K. Debe, Nature 486 (2012) 43-51. DOI:10.1038/nature11115 |
| [3] |
I.M. AlNashef, M.L. Leonard, M.C. Kittle, M.A. Matthews, J.W. Weidner, Electrochem. Solid St 4 (2001) D16-D18. DOI:10.1149/1.1406997 |
| [4] |
M. Hayyan, F.S. Mjalli, M.A. Hashim, I.M. AlNashef, X.M. Tan, J. Electroanal. Chem. 657 (2011) 150-157. DOI:10.1016/j.jelechem.2011.04.008 |
| [5] |
J. Herranz, A. Garsuch, H.A. Gasteiger, J. Phys. Chem. C 116 (2012) 19084-19094. DOI:10.1021/jp304277z |
| [6] |
E.E. Switzer, R. Zeller, Q. Chen, et al., J. Phys. Chem. C 117 (2013) 8683-8690. DOI:10.1021/jp400845u |
| [7] |
X.Z. Yuan, V. Alzate, Z. Xie, et al., J. Electrochem. Soc. 161 (2014) A458-A466. DOI:10.1149/2.010404jes |
| [8] |
L. Xiong, E.O. Barnes, R.G. Compton, Sensor. Actuat. B:Chem. 200 (2014) 157-166. DOI:10.1016/j.snb.2014.04.052 |
| [9] |
X.Z. Yuan, V. Alzate, Z. Xie, D.G. Ivey, W. Qu, J. Electrochem. Soc. 161 (2014) A451-A457. DOI:10.1149/2.008404jes |
| [10] |
J. Schnaidt, T.L. Nguyen, Z. Jusys, R.J. Behm, Electrochim. Acta 299 (2019) 372-377. DOI:10.1016/j.electacta.2018.12.159 |
| [11] |
H.M. Jiang, L. Wang, L.L. Qiao, et al., Int. J. Electrochem. Sci. 14 (2019) 2645-2654. |
| [12] |
L. Wang, S. Liu, H. Jiang, et al., J. Electrochem. Soc. 165 (2018) H705-H710. DOI:10.1149/2.0801811jes |
| [13] |
III W.P. Mounfield, A. Garg, Y. Shao-Horn, Y. Román-Leshkov, Chem 4 (2018) 18-19. DOI:10.1016/j.chempr.2017.12.015 |
| [14] |
X. Huang, Z. Zhao, L. Cao, et al., Science 348 (2015) 1230-1234. DOI:10.1126/science.aaa8765 |
| [15] |
A.A. Gewirth, J.A. Varnell, A.M. DiAscro, Chem. Rev. 118 (2018) 2313-2339. DOI:10.1021/acs.chemrev.7b00335 |
| [16] |
Y. Chen, I. Kone, Y. Gong, et al., Carbon 152 (2019) 325-334. DOI:10.1016/j.carbon.2019.06.026 |
| [17] |
G. Wu, K.L. More, C.M. Johnston, P. Zelenay, Science 332 (6028) 443-447. |
| [18] |
Y. Chen, S. Ji, Y. Wang, et al., Angew. Chem. Int. Ed. 56 (2017) 6937-6941. DOI:10.1002/anie.201702473 |
| [19] |
K. Gong, F. Du, Z. Xia, M. Durstock, L. Dai, Science 323 (2009) 760-764. DOI:10.1126/science.1168049 |
| [20] |
L. Yang, X. Zeng, W. Wang, D. Cao, Adv. Funct. Mater. 28 (2018) 1704537. DOI:10.1002/adfm.201704537 |
| [21] |
M. Tahir, L. Pan, F. Idrees, et al., Nano Energy 37 (2017) 136-157. DOI:10.1016/j.nanoen.2017.05.022 |
| [22] |
K.A. Stoerzinger, L. Qiao, M.D. Biegalski, Y. Shao-Horn, J. Phys. Chem. Lett. 5 (2014) 1636-1641. DOI:10.1021/jz500610u |
| [23] |
Z.F. Huang, J. Song, Y. Du, et al., Nat. Energy 4 (2019) 329-338. DOI:10.1038/s41560-019-0355-9 |
| [24] |
Y. Lee, J. Suntivich, K.J. May, E.E. Perry, Y. Shao-Horn, J. Phys. Chem. Lett. 3 (2012) 399-404. DOI:10.1021/jz2016507 |
| [25] |
Y. Yan, B.Y. Xia, B. Zhao, X. Wang, J. Mater. Chem. A 4 (2016) 17587-17603. DOI:10.1039/C6TA08075H |
| [26] |
R. Zhang, Y.C. Zhang, L. Pan, et al., ACS Catal. 8 (2018) 3803-3811. DOI:10.1021/acscatal.8b01046 |
| [27] |
D. Yan, Y. Li, J. Huo, et al., Adv. Mater. 29 (2017) 1606459. DOI:10.1002/adma.201606459 |
| [28] |
F. Garcés-Pineda, M. Blasco-Ahicart, D. Nieto-Castro, N. López, J.R. GalánMascarós, Nat. Energy 4 (2019) 519-525. DOI:10.1038/s41560-019-0404-4 |
| [29] |
A.B. Laursen, A.S. Varela, F. Dionigi, et al., J. Chem. Educ. 89 (2012) 1595-1599. DOI:10.1021/ed200818t |
| [30] |
E. Skúlason, V. Tripkovic, M.E. Björketun, et al., J. Phys. Chem. C 114 (2010) 18182-18197. DOI:10.1021/jp1048887 |
| [31] |
N. Danilovic, R. Subbaraman, D. Strmcnik, V. Stamenkovic, N. Markovic, J. Serb. Chem. Soc. 78 (2013) 2007-2015. DOI:10.2298/JSC131118136D |
| [32] |
A. Wang, J. Li, T. Zhang, Nat. Rev. Chem. 2 (2018) 65-81. DOI:10.1038/s41570-018-0010-1 |
| [33] |
D. Voiry, J. Yang, M. Chhowalla, Adv. Mater. 28 (2016) 6197-6206. DOI:10.1002/adma.201505597 |
| [34] |
C. Hu, L. Dai, Adv. Mater. 31 (2019) 1804672. DOI:10.1002/adma.201804672 |
| [35] |
J. Wei, M. Zhou, A. Long, et al., Nanomicro Lett. 10 (2018) 75. |
| [36] |
N. Danilovic, R. Subbaraman, D. Strmcnik, et al., Angew. Chem. Int. Ed. 51 (2012) 12495-12498. DOI:10.1002/anie.201204842 |
| [37] |
M. Lao, K. Rui, G. Zhao, et al., Angew. Chem. Int. Ed. 58 (2019) 5432-5437. DOI:10.1002/anie.201901010 |
| [38] |
G.Q. Zhao, K. Rui, S.X. Dou, W.P. Sun, Adv. Funct. Mater. 28 (2018) 1803291. DOI:10.1002/adfm.201803291 |
| [39] |
R.K. Pachauri, M.R. Allen, V.R. Barros (Eds.), et al., Climate Change 2014: Synthesis Report. Contribution of Working Groups Ⅰ, Ⅱ and Ⅲ to the fifth assessment report of the Intergovernmental Panel on Climate Change, IPCC, Geneva, 2014, pp. 151.
|
| [40] |
T. Zheng, K. Jiang, H. Wang, Adv. Mater. 30 (2018) 1802066. DOI:10.1002/adma.201802066 |
| [41] |
T. Cheng, H. Xiao, W.A. Goddard III, J. Am. Chem. Soc. 138 (2016) 13802-13805. DOI:10.1021/jacs.6b08534 |
| [42] |
W. Zhu, Y.J. Zhang, H. Zhang, et al., J. Am. Chem. Soc. 136 (2014) 16132-16135. DOI:10.1021/ja5095099 |
| [43] |
N. Han, Y. Wang, H. Yang, et al., Nat. Commun. 9 (2018) 1320. DOI:10.1038/s41467-018-03712-z |
| [44] |
H. Xiao, T. Cheng, W.A. Goddard 3rd, R. Sundararaman, J. Am. Chem. Soc. 138 (2016) 483-486. DOI:10.1021/jacs.5b11390 |
| [45] |
T. Cheng, H. Xiao, W.A. Goddard, P. Natl. Acad. Sci. U. S. A. 114 (2017) 1795-1800. DOI:10.1073/pnas.1612106114 |
| [46] |
C.T. Dinh, T. Burdyny, M.G. Kibria, et al., Science 360 (2018) 783-787. DOI:10.1126/science.aas9100 |
| [47] |
S. Lee, G. Park, J. Lee, ACS Catal. 7 (2017) 8594-8604. DOI:10.1021/acscatal.7b02822 |
| [48] |
Y.C. Li, Z. Wang, T. Yuan, et al., J. Am. Chem. Soc. 141 (2019) 8584-8591. DOI:10.1021/jacs.9b02945 |
| [49] |
Z.W. Seh, J. Kibsgaard, C.F. Dickens, et al., Science 355 (2017) eaad4998. DOI:10.1126/science.aad4998 |
| [50] |
R. Zhao, H. Xie, L. Chang, et al., Energy Chem. (2019) 100011. |
| [51] |
L.C. Seefeldt, B.M. Hoffman, D.R. Dean, Annu. Rev. Biochem. 78 (2009) 701-722. DOI:10.1146/annurev.biochem.78.070907.103812 |
| [52] |
R.R. Eady, Chem. Rev. 96 (1996) 3013-3030. DOI:10.1021/cr950057h |
| [53] |
L. Zhang, X. Ji, X. Ren, et al., Adv. Mater. 30 (2018) e1800191. DOI:10.1002/adma.201800191 |
| [54] |
X. Li, T. Li, Y. Ma, et al., Adv. Energy Mater. 8 (2018) 1801357. |
| [55] |
Y. Liu, M. Han, Q. Xiong, et al., Adv. Energy Mater. 9 (2019) 1803935. |
| [56] |
J. Han, X. Ji, X. Ren, et al., J. Mater. Chem. A 6 (2018) 12974-12977. DOI:10.1039/C8TA03974G |
| [57] |
X. Ren, G. Cui, L. Chen, et al., Chem. Commun. (Camb) 54 (2018) 8474-8477. DOI:10.1039/C8CC03627F |
| [58] |
X. Ren, J. Zhao, Q. Wei, et al., ACS Central Sci. 5 (2019) 116-121. DOI:10.1021/acscentsci.8b00734 |
| [59] |
S. Licht, B. Cui, B. Wang, et al., Science 345 (2014) 637-640. DOI:10.1126/science.1254234 |
| [60] |
S. Chen, S. Perathoner, C. Ampelli, et al., Angew. Chem. Int. Ed. 56 (2017) 2699-2703. DOI:10.1002/anie.201609533 |
| [61] |
Q. Liu, X. Zhang, B. Zhang, et al., Nanoscale 10 (2018) 14386-14389. DOI:10.1039/C8NR04524K |
| [62] |
X. Zhu, Z. Liu, Q. Liu, et al., Chem. Comm. 54 (2018) 11332-11335. DOI:10.1039/C8CC06366D |
| [63] |
X. Zhu, Z. Liu, H. Wang, et al., Chem. Commun. 55 (2019) 3987-3990. DOI:10.1039/C9CC00647H |
| [64] |
X. Zhao, X. Lan, D. Yu, et al., Chem. Commun. 54 (2018) 13010-13013. DOI:10.1039/C8CC08045C |
| [65] |
Y. Abghoui, A.L. Garden, J.G. Howalt, T. Vegge, E. Skúlason, ACS Catal. 6 (2015) 635-646. |
| [66] |
R. Zhang, Y. Zhang, X. Ren, et al., ACS Sustain. Chem. Eng. 6 (2018) 9545-9549. DOI:10.1021/acssuschemeng.8b01261 |
| [67] |
R. Zhang, J. Han, B. Zheng, et al., Inorg. Chem. Front. 6 (2019) 391-395. DOI:10.1039/C8QI01145A |
| [68] |
R. Zhang, H. Guo, L. Yang, et al., Chem. Electro. Chem. 6 (2019) 1014-1018. |
| [69] |
Z. Wang, F. Gong, L. Zhang, et al., Adv. Sci. 6 (2019) 1801182. DOI:10.1002/advs.201801182 |
| [70] |
L. Yang, T. Wu, R. Zhang, et al., Nanoscale 11 (2019) 1555-1562. DOI:10.1039/C8NR09564G |
| [71] |
Y. Zhang, W. Qiu, Y. Ma, et al., ACS Catal. 8 (2018) 8540-8544. DOI:10.1021/acscatal.8b02311 |
| [72] |
J. Han, Z. Liu, Y. Ma, et al., Nano Energy 52 (2018) 264-270. DOI:10.1016/j.nanoen.2018.07.045 |
| [73] |
W. Kong, R. Zhang, X. Zhang, et al., Nanoscale 11 (2019) 19274-19277. DOI:10.1039/C9NR03678D |
| [74] |
K. Chu, Y.P. Liu, Y.B. Li, H. Zhang, Y. Tian, J. Mate. Chem. A 7 (2019) 4389-4394. DOI:10.1039/C9TA00016J |
| [75] |
L. Zhang, X. Ren, Y. Luo, et al., Chem. Commun. 54 (2018) 12966-12969. DOI:10.1039/C8CC06524A |
| [76] |
W. Qiu, X.Y. Xie, J. Qiu, et al., Nat. Commun. 9 (2018) 3485. DOI:10.1038/s41467-018-05758-5 |
| [77] |
L. Xia, X. Wu, Y. Wang, et al., Small Methods 3 (2019) 1800251. DOI:10.1002/smtd.201800251 |
| [78] |
L. Xia, J.J. Yang, H.B. Wang, et al., Chem. Commun. 55 (2019) 3371-3374. DOI:10.1039/C9CC00602H |
| [79] |
H. Chen, X. Zhu, H. Huang, et al., Chem. Commun. 55 (2019) 3152-3155. DOI:10.1039/C9CC00461K |
| [80] |
X. Yu, P. Han, Z. Wei, et al., Joule 2 (2018) 1610-1622. DOI:10.1016/j.joule.2018.06.007 |
| [81] |
T. Wang, L. Xia, J.J. Yang, et al., Chem. Commun. 55 (2019) 7502-7505. DOI:10.1039/C9CC01999E |
| [82] |
J. Zhao, B. Wang, Q. Zhou, et al., Chem. Commun. 55 (2019) 4997-5000. DOI:10.1039/C9CC00726A |
| [83] |
Y. Song, T. Wang, J. Sun, et al., ACS Sustain. Chem. Eng. 49 (2019) 5654-5662. |
| [84] |
X. Zhang, T. Wu, H. Wang, et al., ACS Catal. 9 (2019) 4609-4615. DOI:10.1021/acscatal.8b05134 |
| [85] |
L. Zhang, L.X. Ding, G.F. Chen, X. Yang, H. Wang, Angew. Chem. 131 (2019) 2638-2642. DOI:10.1002/ange.201813174 |
| [86] |
X. Zhu, T. Wu, L. Ji, et al., J. Mater. Chem. A 7 (2019) 16117-16121. DOI:10.1039/C9TA05016G |
| [87] |
Y. Zhang, H.T. Du, Y.J. Ma, et al., Nano Res. 12 (2019) 919-924. DOI:10.1007/s12274-019-2323-x |
| [88] |
C. Lv, Y. Qian, C. Yan, et al., Angew. Chem. Int. Ed. 57 (2018) 10246-10250. DOI:10.1002/anie.201806386 |
| [89] |
S. Fukuzumi, Y.M. Lee, W. Nam, Chemistry 24 (2018) 5016-5031. DOI:10.1002/chem.201704512 |
| [90] |
M. Neumann-Spallart, K. Kalyanasundaram, J. Phys. Chem. 86 (1982) 2681-2690. DOI:10.1021/j100211a025 |
| [91] |
P. Lianos, Appl. Catal. B:Environ. 210 (2017) 235-254. DOI:10.1016/j.apcatb.2017.03.067 |
| [92] |
H. Selcuk, W. Zaltner, J. Sene, M. Bekbolet, M. Anderson, J. Appl. Electrochem. 34 (2004) 653-658. DOI:10.1023/B:JACH.0000021931.36151.54 |
| [93] |
K. Mase, M. Yoneda, Y. Yamada, S. Fukuzumi, ACS Energy Lett. 1 (2016) 913-919. DOI:10.1021/acsenergylett.6b00415 |
| [94] |
M. Gryszel, A. Markov, M. Vagin, E.D. Głowacki, J. Mater. Chem. A 6 (2018) 24709-24716. DOI:10.1039/C8TA08151D |
| [95] |
O. Jung, M.L. Pegis, Z. Wang, et al., J. Am. Chem. Soc. 140 (2018) 4079-4084. DOI:10.1021/jacs.8b00015 |
| [96] |
A.S. Konev, M.Y. Kayumov, M.P. Karushev, et al., ChemElectroChem 5 (2018) 3138-3142. DOI:10.1002/celc.201800846 |
| [97] |
J. Sun, Y. Yu, A.E. Curtze, X. Liang, Y. Wu, Chem. Sci. 10 (2019) 5519-5527. DOI:10.1039/C9SC01626K |
| [98] |
M. Jakešová, D.H. Apaydin, M. Sytnyk, et al., Adv. Funct. Mater. 26 (2016) 5248-5254. DOI:10.1002/adfm.201601946 |
| [99] |
P. Luan, J. Zhang, ChemElectroChem 6 (2019) 3227-3243. DOI:10.1002/celc.201900398 |
| [100] |
D. Raptis, V. Dracopoulos, P. Lianos, J. Hazard. Mater. 333 (2017) 259-264. DOI:10.1016/j.jhazmat.2017.03.044 |
| [101] |
E. Kalamaras, V. Dracopoulos, L. Sygellou, P. Lianos, Chem. -Eng. J. 295 (2016) 288-294. DOI:10.1016/j.cej.2016.03.062 |
| [102] |
I. Papagiannis, E. Doukas, A. Kalarakis, G. Avgouropoulos, P. Lianos, Catalysts 9 (2019) 243. DOI:10.3390/catal9030243 |
| [103] |
O. Monfort, S. Sfaelou, L. Satrapinskyy, et al., Catal. Today 280 (2017) 51-57. DOI:10.1016/j.cattod.2016.07.006 |
| [104] |
P. Panagiotopoulou, M. Antoniadou, D.I. Kondarides, P. Lianos, Appl. Catal. B:Environ. 100 (2010) 124-132. DOI:10.1016/j.apcatb.2010.07.021 |
| [105] |
I. Tantis, M. Antonopoulou, I. Konstantinou, P. Lianos, J. Photoch. Photobio. A 317 (2016) 100-107. DOI:10.1016/j.jphotochem.2015.11.013 |
| [106] |
K. Mase, M. Yoneda, Y. Yamada, S. Fukuzumi, Nat. Commun. 7 (2016) 11470. DOI:10.1038/ncomms11470 |
| [107] |
M. Grätzel, Nature 414 (2001) 338-344. DOI:10.1038/35104607 |
| [108] |
A. Fujishima, K. Honda, Nature 238 (1972) 37-38. DOI:10.1038/238037a0 |
| [109] |
S. Sakthivel, M. Janczarek, H. Kisch, J. Phys. Chem. B 108 (2004) 19384-19387. DOI:10.1021/jp046857q |
| [110] |
S. Hoang, S. Guo, N.T. Hahn, A.J. Bard, C.B. Mullins, Nano Lett. 12 (2011) 26-32. |
| [111] |
Z.F. Hu, J.C. Yu, T. Ming, J.F. Wang, Appl. Catal. B-Environ. 168 (2015) 483-489. |
| [112] |
E.S. Kim, N. Nishimura, G. Magesh, et al., J.Am.Chem.Soc. 135 (2013) 5375-5383. DOI:10.1021/ja308723w |
| [113] |
P.M. Rao, L. Cai, C. Liu, et al., Nano Lett. 14 (2014) 1099-1105. DOI:10.1021/nl500022z |
| [114] |
Y. Hou, F. Zuo, A. Dagg, P. Feng, Angew. Chem. Int. Ed. 52 (2013) 1248-1252. DOI:10.1002/anie.201207578 |
| [115] |
Z. Hu, M. Xu, Z. Shen, C.Y. Jimmy, J. Mater. Chem. A 3 (2015) 14046-14053. DOI:10.1039/C5TA02528A |
| [116] |
L. Pan, S. Wang, J. Xie, et al., Nano Energy 28 (2016) 296-303. DOI:10.1016/j.nanoen.2016.08.054 |
| [117] |
Z. Hu, Z. Shen, C.Y. Jimmy, F. Cheng, Appl. Catal. B:Environ. 203 (2017) 829-838. DOI:10.1016/j.apcatb.2016.10.079 |
| [118] |
B. Klahr, S. Gimenez, F. Fabregat-Santiago, J. Bisquert, T.W. Hamann, J. Am. Chem. Soc. 134 (2012) 16693-16700. DOI:10.1021/ja306427f |
| [119] |
D.K. Zhong, D.R. Gamelin, J. Am. Chem. Soc. 132 (2010) 4202-4207. DOI:10.1021/ja908730h |
| [120] |
P. Tang, H. Xie, C. Ros, et al., Energy Environ. Sci. 10 (2017) 2124-2136. DOI:10.1039/C7EE01475A |
| [121] |
Z. Hu, Z. Shen, J.C. Yu, Chem. Mater. 28 (2016) 564-572. DOI:10.1021/acs.chemmater.5b04058 |
| [122] |
C.F. Shih, T. Zhang, J. Li, C. Bai, Joule (2018) 1925-1949. |
| [123] |
B.H. Farnum, K.R. Wee, T.J. Meyer, Nat. Chem. 8 (2016) 845-852. DOI:10.1038/nchem.2536 |
| [124] |
G. Sahara, R. Abe, M. Higashi, et al., Chem. Commun. 51 (2015) 10722-10725. DOI:10.1039/C5CC02403J |
| [125] |
D. Wang, Y. Wang, M.D. Brady, et al., Chem. Sci. 10 (2019) 4436-4444. DOI:10.1039/C8SC03316A |
| [126] |
J.J. Leung, J. Warnan, K.H. Ly, et al., Nat. Catal. 2 (2019) 354-365. DOI:10.1038/s41929-019-0254-2 |
| [127] |
H. Takeda, H. Kamiyama, K. Okamoto, et al., J. Am. Chem. Soc. 140 (2018) 17241-17254. DOI:10.1021/jacs.8b10619 |
| [128] |
J. Messinger, O. Ishitani, D. Wang, Sustain. Energ. Fuels 2 (2018) 1891-1892. DOI:10.1039/C8SE90049C |
| [129] |
X. Jiao, Z. Chen, X. Li, et al., J. Am. Chem. Soc. 139 (2017) 7586-7594. DOI:10.1021/jacs.7b02290 |
| [130] |
L. D'Amario, J. Föhlinger, G. Boschloo, L. Hammarström, Chem. Sci. 9 (2018) 223-230. DOI:10.1039/C7SC03442C |
| [131] |
H. Zhang, H. Liu, Z. Tian, et al., Nat. Nanotechnol. 13 (2018) 900-905. DOI:10.1038/s41565-018-0267-z |
| [132] |
K.P. Sokol, W.E. Robinson, A.R. Oliveira, et al., J. Am. Chem. Soc. 140 (2018) 16418-16422. DOI:10.1021/jacs.8b10247 |
| [133] |
M. Schreier, F. Héroguel, L. Steier, et al., Nat. Energy 2 (2017) 17087. DOI:10.1038/nenergy.2017.87 |
| [134] |
Y. Wang, J. Liu, Y. Wang, Y. Wang, G. Zheng, Nat. Commun. 9 (2018) 5003. DOI:10.1038/s41467-018-07380-x |
| [135] |
K.C. Christoforidis, P. Fornasiero, ChemCatChem 9 (2017) 1523-1544. DOI:10.1002/cctc.201601659 |
| [136] |
S. Kampouri, K.C. Stylianou, ACS Catal. 9 (2019) 4247-4270. DOI:10.1021/acscatal.9b00332 |
| [137] |
J. Schneider, D.W. Bahnemann, J. Phys. Chem. Lett. 4 (2013) 3479-3483. DOI:10.1021/jz4018199 |
| [138] |
B. Wang, S. Shen, S.S. Mao, J. Materiomics 3 (2017) 96-111. DOI:10.1016/j.jmat.2017.02.001 |
| [139] |
Y. Zhang, J. Gao, Z. Chen, J. Colloid Interf. Sci. 535 (2019) 331-340. DOI:10.1016/j.jcis.2018.10.012 |
| [140] |
S. Cao, L. Piao, X. Chen, Trends in Chemistry (2019), doi: http://dx.doi.org/10.1016/j.trechm.2019.06.009.
|
| [141] |
J. Fu, Q. Xu, J. Low, C. Jiang, J. Yu, Appl. Catal. B:Environ. 243 (2019) 556-565. DOI:10.1016/j.apcatb.2018.11.011 |
| [142] |
Y. Si, S. Cao, Z. Wu, et al., Appl. Catal. B:Environ. 220 (2018) 471-476. DOI:10.1016/j.apcatb.2017.08.024 |
| [143] |
X. Gao, J. Feng, D. Su, et al., Nano Energy 59 (2019) 598-609. DOI:10.1016/j.nanoen.2019.03.016 |
| [144] |
Y. Zhuang, Y. Liu, X. Meng, Appl. Surf. Sci. 496 (2019) 143647. DOI:10.1016/j.apsusc.2019.143647 |
| [145] |
A.R. Singh, B.A. Rohr, M.J. Statt, et al., ACS Catalysis (2019) 8316-8324. |
| [146] |
J. Li, H. Li, G. Zhan, L. Zhang, Acc Chem Res 50 (2017) 112-121. DOI:10.1021/acs.accounts.6b00523 |
| [147] |
H. Li, J. Shang, Z. Ai, L. Zhang, J. Am. Chem. Soc. 137 (2015) 6393-6399. DOI:10.1021/jacs.5b03105 |
| [148] |
G.N. Schrauzer, T.D. Guth, J. Am. Chem. Soc. 99 (1977) 7189-7193. DOI:10.1021/ja00464a015 |
| [149] |
X. Chen, N. Li, Z. Kong, W.J. Ong, X. Zhao, Mater. Horiz. 5 (2018) 9-27. DOI:10.1039/C7MH00557A |
| [150] |
Q. Zhang, S. Hu, Z. Fan, et al., Dalton T. 45 (2016) 3497-3505. DOI:10.1039/C5DT04901F |
| [151] |
T.A. Bu, Y.C. Hao, W.Y. Gao, et al., Nanoscale 11 (2019) 10072-10079. DOI:10.1039/C9NR02502B |
| [152] |
T. Oshikiri, K. Ueno, H. Misawa, Angew. Chem. Int. Ed. 53 (2014) 9802-9805. DOI:10.1002/anie.201404748 |
| [153] |
M. Ali, F. Zhou, K. Chen, et al., Nat. Commun. 7 (2016) 11335. DOI:10.1038/ncomms11335 |
| [154] |
M. Li, H. Huang, J. Low, et al., Small Methods 3 (2019) 1800388. DOI:10.1002/smtd.201800388 |
| [155] |
A. Tsuneto, A. Kudo, T. Sakata, J. Electroanal. Chem. 367 (1994) 183-188. DOI:10.1016/0022-0728(93)03025-K |
| [156] |
N. Lazouski, Z.J. Schiffer, K. Williams, K. Manthiram, Joule 3 (2019) 1127-1139. DOI:10.1016/j.joule.2019.02.003 |
| [157] |
Y.C. Hao, Y. Guo, L.W. Chen, et al., Nat. Catal. 2 (2019) 448. DOI:10.1038/s41929-019-0241-7 |
| [158] |
F. Zhou, L.M. Azofra, M. Ali, et al., Energy Environ. Sci. 10 (2017) 2516-2520. DOI:10.1039/C7EE02716H |
| [159] |
J. Zheng, Y. Lyu, M. Qiao, et al., Chem 5 (2019) 617-633. DOI:10.1016/j.chempr.2018.12.003 |
| [160] |
Y. Nishibayashi, Inorg. Chem. 54 (2015) 9234-9247. DOI:10.1021/acs.inorgchem.5b00881 |
| [161] |
S. Hu, W. Zhang, J. Bai, et al., RSC Adv. 6 (2016) 25695-25702. DOI:10.1039/C5RA28123G |
| [162] |
D.L. Ashford, M.K. Gish, A.K. Vannucci, et al., Chem. Rev. 115 (2015) 13006-13049. DOI:10.1021/acs.chemrev.5b00229 |
| [163] |
X. Zhao, F. Yin, N. Liu, et al., J. Mater. Sci. 52 (2017) 10175-10185. DOI:10.1007/s10853-017-1176-5 |
| [164] |
Y. Qiu, X. Peng, F. Lü, et al., Chem. -Asian J. 14 (2019) 2770-2779. |
| [165] |
S. Sun, Q. An, W. Wang, et al., J. Mater. Chem. A 5 (2017) 201-209. DOI:10.1039/C6TA09275F |
| [166] |
K. Tennakone, C. Thaminimulla, W. Kiridena, Langmuir 9 (1993) 723-726. DOI:10.1021/la00027a019 |
| [167] |
S.Z. Andersen, V. Čolić, S. Yang, et al., Nature 570 (2019) 504-508. DOI:10.1038/s41586-019-1260-x |
| [168] |
K. Li, B. Peng, T. Peng, ACS Catal. 6 (2016) 7485-7527. DOI:10.1021/acscatal.6b02089 |
| [169] |
J.K. Stolarczyk, S. Bhattacharyya, L. Polavarapu, J. Feldmann, ACS Catal. 8 (2018) 3602-3635. DOI:10.1021/acscatal.8b00791 |
| [170] |
Y. Zheng, A. Vasileff, X. Zhou, et al., J. Am. Chem. Soc. 141 (2019) 7646-7659. DOI:10.1021/jacs.9b02124 |
| [171] |
B. Han, X. Ou, Z. Deng, et al., Angew. Chem. Int. Ed. 57 (2018) 16811-16815. DOI:10.1002/anie.201811545 |
| [172] |
X. Li, J. Yu, M. Jaroniec, X. Chen, Chem. Rev. 119 (2019) 3962-4179. DOI:10.1021/acs.chemrev.8b00400 |
| [173] |
X. Su, X.F. Yang, Y. Huang, B. Liu, T. Zhang, Accounts Chem. Res. 52 (2018) 656-664. |
| [174] |
H. Wang, X. Zhang, Y. Xie, Mat. Sci. Eng. R 130 (2018) 1-39. DOI:10.1016/j.mser.2018.04.002 |
| [175] |
J. Ran, M. Jaroniec, S.Z. Qiao, Adv. Mater. 30 (2018) 1704649. DOI:10.1002/adma.201704649 |
| [176] |
S. Xu, E.A. Carter, Chem. Rev. 119 (2018) 6631-6669. |