 2019, Vol. 30
2019, Vol. 30 


b Key Laboratory of Smart Drug Delivery(Fudan University), Ministry of Education, Shanghai 201203, China
The self-assembly of amphiphilic polymers with hydrophilic and hydrophobic units has emerged as a promising approach to create functional materials [1-4]. However, the self-assembly research was dominated by the amphiphilic block polymers [5-8]. Recently, it has been documented that the amphiphilic random copolymers also exhibit excellent self-folding behavior [9-11]. For examples, Feng's group synthesized the amphiphilic random terpolymer poly(AcTreMA-co-PEGMA-co-13FOMA) with fluorinated PEG/ trehalose and investigated the effect of hydrophilic and hydrophobic units on the self-assembly [12]. Hattori's group developed a general self-assembly system to nanostructured materials in both solid and solution using common amphiphilic random copolymers with hydrophilic poly(ethylene glycol) (PEG) and hydrophobic crystalline octadecyl pendants [13].
The stimulus-responsive polymers can adapt to the surrounding environment, such as changes in temperature, chemicalcomposition or applied mechanical force, or changes triggered externally by light or exposure to electric and magnetic fields [14-18]. The increasing number of burgeoning applications of these materials which rely on very distinct responses on a macroscopic and/or microscopic level. This can be achieved by processing stimulus-responsive polymers into nanostructured materials. It is well known that amphiphilic polymers could self-assemble into nanoparticles in selective solvents [19-25]. For examples, Zhang's group synthesized the GSH-responsive polymer (mPEG-SS-CPT) and the enzyme-responsive polymer (PBA-PEG-Azo-PCL). The mixture of the polymeric prodrug and the enzyme-responsive copolymer could self-assemble into micelles [26].Thedual-responsive copolymers(PEG-SS-COOH) with GSH-sensitive disulfide bonds and pH-responsive carboxylic acid groups were synthesized by BEng's group. And the block copolymer could self-assemble into micelles in selective solvents [27]. Hence, the stimulus-responsive self-assembly polymers were playing an increasingly important role in a wide variety of applications.
Aliphatic polycarbonates (APCs) have great potential application due to their unique functionality [28, 29]. Advanced work was reported that the stimulus-responsive self-assembly polycarbonate was prepared by introducing hydrophilic polymers into hydrophobic polycarbonate [30, 31]. For examples, Yang's group synthesized the amphiphilic copolymers mPEG-b-CMP45 with the hydrophilic mPEG and the functional selenium-containing hydrophobic polycarbonate, which could self-assemble and is response to ROS [32]. Ganivada's group designed and synthesized the Bt- (PE—PA—PC)-DRoxm containing PEG, doxorubicin (DOX) and biotin receptor conjugate. Among the three functionalities, PEG was attached to end groups as hydrophilic units and doxorubicin was grafted as the pendent motif to the poly(lactide-carbonate) backbone [33]. Moreover, to the best of our knowledge, there was no research on GSH/pH dual-responsive copolymers prepared entirely from poly-carbonate with the function of hydrophilic and hydrophobic, which can self-assemble into micelles in water so far.

|
Download:
|
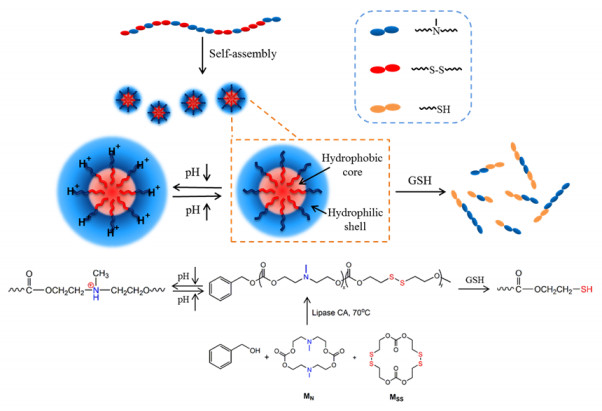
| Scheme 1. Synthesis of poly(MN-co-MSS) copolymer by lipase-catalyzed ring-opening polymerization and the reduction and pH-response behavior of poly(MN-co-MSS). | |
{kind=link}
Here, a novel dual-responsive polycarbonate assemblies were constructed by the functionalized carbonate monomers based on our previous study (the carbonate monomers structures were shown in Figs. S1 and S2 in Supporting information). The dualresponsive tertiary amine/ disulfide functionalized polycarbonate (poly(MN-co-MSS)) with tertiary amine and disulfide groups on the backbone via an enzyme-catalyzed ring-opening copolymerization. It should be noted that the tertiary amino groups introduced hydrophilicity into the hydrophobic polycarbonate, and consequently the polycarbonate with hydrophilic/hydrophobic can easily self-assemble into micelle-like aggregates in aqueous solution. Further, the disulfide groups as well as tertiary amine groups endowed the micelles with rich GSH and pH responsiveness respectively. The results highlighted a facile synthesis of the environment-responsive polymers and provided a novel GSH/pH responsive material platform for further application.
As shown in Scheme 1, the poly(MN-co-MSS) was prepared via the ring opening copolymerization of MN and MSS, which was initiated with the hydroxyl group of benzyl alcohol and catalyzed by lipase CA(N-435). The two monomers (MN and MSS) were randomly distributed on the backbone chain, and the mechanism of the self-assembly and dual-responsiveness of poly(MN-co-MSS) was proposed. The random copolymer poly(MN-co-MSS) selfassemble into micelle-like aggregates with hydrophobic core and hydrophilic shell. And the micelles size increased under the condition of low pH because of the charge repulsion of the protonated tertiary amino groups. In the reducing environment (GSH), the disulfide bonds were reduced to hydrophilic sulfhydryl groups and the micellar structures were disassembled into a disordered polymer chains.
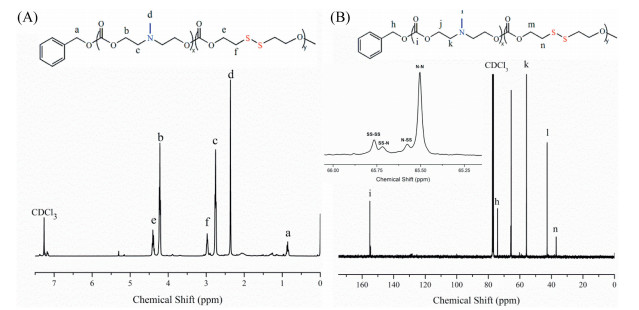
In order to gain insight into their chemical structure, poly (MN-co-MSS) were subjected to NMR and FTIR measurements. The cyclic functional carbonate dimers MN and MSS were synthesized using the macro-ring closure of the functional diol (N-methyl diethanolamine and disulfide ethanol) as we reported before [34]. It was obvious from Fig. 1A that the peak a at 5.42 ppm was assigned to the protons of BnOH. The peak b at 4.24 ppm and the peak c at 2.77 ppm were attributed to the methylene protons (-OCH2CH2N-) respectively. The peak e at 4.40 ppm and the peak f at 2.97 ppm were attributed to the methylene protons (-OCH2CH2SS-) respectively. Besides, the peak d at 2.38 ppm was ascribed to the methyl attached to the nitrogen.

|
Download:
|
| Fig. 1. Typical 1H NMR (A) and 13C NMR spectra (B) of poly(MN-co-MSS). | |
{kind=link}
Furthermore, 13C NMR (Fig. 1B) was employed to confirm the structure and the monomer sequence in the polymer. The peak h at 74 ppm was assigned to the carbon of BnOH (PH-CH2). The peak i at 155 ppm was ascribed to the carbonyl carbon in the polymer chains (-O-C = O). The peak k at 55 ppm and the peak n at 37 ppm were attributed to the carbon near nitrogen (-N-C-) and the carbon near sulfur (-SS-C-) respectively. The four peaks from 65 ppm to 66 ppm were assigned to the carbon near the carbonyl group (-C-O-C = O) respectively, which illustrated the randomness of the polycarbonate segments [34].
In addition, the FTIR spectra of the PN and the poly(MN-MSS) was shown in Fig. S3 (Supporting information). It was observed that the peak at 1741 cm-1 was assigned to the carbonyl groups (-O-C = O). The characteristic peaks at 1400 cm-1 and 526 cm-1 were attributed to -C-N- and -S-S- respectively.
The thermal stability of copolymer poly(MN-co-MSS) was investigated on TGA measurements. As shown in Fig. S4 (Supporting information), the thermal decomposition of poly(MN-co-MSS) presented only one stage starting at 203 ℃, which was due to the decomposition of the carbonate chain. The result illustrated that the copolymer poly(MN-co-MSS) was a homogeneous system. Therefore, we hypothesized that the two monomers arranged randomly on the backbone chain of the copolymer.
In a word, these results indicated the successful synthesis of the poly(MN-co-MSS) copolymers, and the NMR, FTIR and TGA agreed very well.
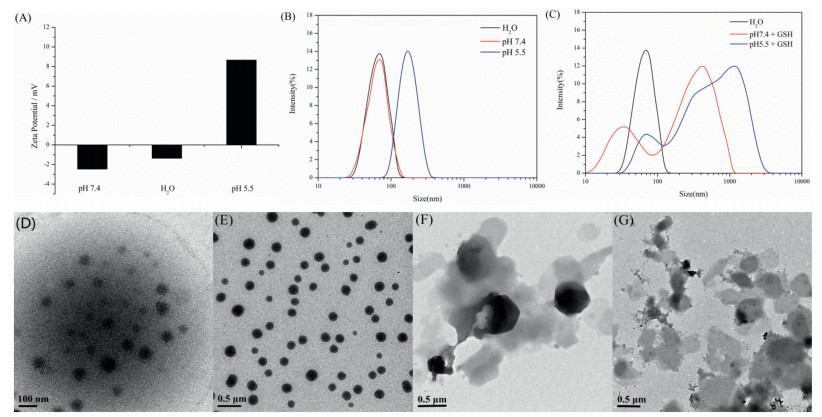
In order to gain insight into the mechanism of the self-assembly and dual-responsiveness, poly(MN-co-MSS) were subjected to NMR, DLS, TEM and GPC measurements. It is generally accepted that disulfide bond can be reduced to a sulfhydryl group [35]. So we were encouraged to investigate the reduction-responsive behavior of the poly(MN-co-MSS). Here, the dual-responsive behaviors of the micelles were further studied using DLS by monitoring the change in the size in response to acid and GSH environments. It was observed from Fig. 2 that the particle size increased at acid or GSH environment for 48 h. As shown in Figs. 2A and B, the pH was tuned at 5.5, the average size was around 170 nm. The reason may be that the easy protonation of poly(MN) core which could facilitate the particles more dispersible due to the charge repulsion and enlarged hydration capacity under the condition of low pH [36]. Upon pH 7.4, the protonation-deprotonation transition of the tertiary amine containing segments led to the stable size (~69 nm) compared with the size in water (~70 nm). The protonation of poly (MN) core was confirmed by the measurement of zeta-potential, which changed from negative (~2.2 mV) to positive (+8.4 mV) because of the positively charged tertiary amine groups.

|
Download:
|
| Fig. 2. Zeta potential and DLS results of the poly(MN-co-MSS) copolymers with (A, B) different pH or (C) different pH and 30 mmol/L GSH at 37 ℃ for 48 h. And TEM images of poly(MN-co-MSS) particles in different solution at 37 ℃. (D) Aqueous solution, (E) pH 5.5, (F) Reduction for 48 h by 30 mmol/L GSH, (G) pH 5.5 and reduction for 48 h by 30 mmol/L GSH. | |
{kind=link}
Also, after the addition of 30 mmol/L GSH at 37 ℃ for 48 h, the particle size increased (Fig. 2C). The GSH could induce the reduction of disulfide to sulfhydryl group which markedly increased the hydrophilicity. Thus the micellar structure destructed and the particle size decreased. Then some of the polymer chains were agglomerated, thus the particle size generally increased [35].
Here, the microscopic images of the micelles were confirmed by TEM. The copolymer poly(MN-co-MSS) firstly self-assemble into micelle-like aggregates in aqueous solution (Fig. 2D). As shown in Fig. 2E, the micelles size increased upon pH 5.5. After reduction for 48 h by 30 mmol/L GSH, the micellar morphologies were destroyed and the polymer chains agglomerated into irregular aggregates (Figs. 2F and G). From the DLS and TEM, it was evolved that the copolymer poly(MN-co-MSS) could selfassemble into micelles, and they would expand and then disassemble after acidification and reduction. The difference in micelles size between DLS and TEM can be attributed to the dehydrated state of the micelles [37].
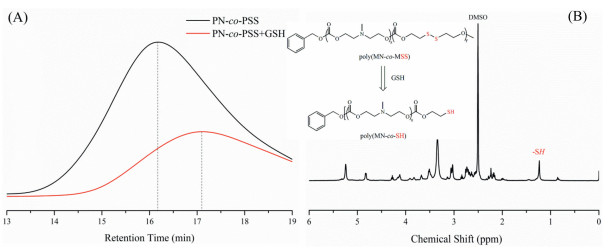
Moreover, GPC and 1H NMR were applied to trace the reduction products. As seen from Fig. 3A, after the reduction of 30 mmol/L GSH for 48 h, the molecular weight was lower and the molecular weight distribution (PDI) became wider, which was due to the formation of corresponding sulfhydryl groups and the disordered aggregation of the broken polymer chains. And it was obvious from Fig. 3B that the peak at 1.32 ppm was assigned to the protons of sulfhydryl (-SH) after reduction.

|
Download:
|
| Fig. 3. GPC trace of poly(MN-co-MSS) before and after reduction with 30 mmol/L GSH for 48 h (A) and reduction-responsive cleavage of the disulfide group in the synthesized polymer poly(MN-co-MSS) and the typical 1H NMR of the poly(MN-co-SH) (B). | |
{kind=link}
In summary, a dual-stimuli-responsive micellar aggregates were obtained by the functionalized polycarbonate itself. The polycarbonate was synthesized via enzyme-catalyzed ring opening copolymerization of the functionalized carbonate monomers bearing different groups. The tertiary amine and disulfide groups endowed the copolymers with rich pH and reductive responsiveness by the protonation-deprotonation process and the reduction of the disulfide groups to hydrophilic sulfydryl groups, and the responsive process was clarified in detail. Thus, we believe that the poly(MN-co-MSS) copolymer could broaden the stimulus responsiveness polymers' family and would have great potential as nanocarriers for cancer treatment in the future.
AcknowledgementsThis work was supported by the National Natural Science Foundation of China (Nos. 51873062, 81673018), Shanghai Natural Science Foundation (No.17ZR1407100), School of Pharmacy, Fudan University & The Open Project Program of Key Lab of Smart Drug Delivery (Fudan University, Nos. SDD2017-04, SDD2019-01).
Appendix A. Supplementary dataSupplementary material related to this article can be found, inthe online version, at doi:https://doi.org/10.1016/j.cclet.2019.05.017.
| [1] |
Z.G. Xu, S.Y. Liu, Y.J. Kang, M.F. Wang, ACS Biomater. Sci. Eng. 1 (2015) 585-592. DOI:10.1021/acsbiomaterials.5b00119 |
| [2] |
L.N. Gu, Z. Shen, S. Zhang, et al., Macromolecules 40 (2007) 4486-4493. DOI:10.1021/ma062083v |
| [3] |
W.H. Qian, T. Song, M. Ye, et al., Polym. Chem. 8 (2017) 4098. DOI:10.1039/C7PY00762K |
| [4] |
J. Zhu, H. Han, F.X. Li, et al., J. Colloid Interface Sci. 540 (2019) 634-646. DOI:10.1016/j.jcis.2018.12.091 |
| [5] |
S.K. Ahn, J. Nam, J.H. Zhu, E. Lee, S.M. Kilbey, Polym. Chem. 9 (2018) 3279-3286. DOI:10.1039/C8PY00627J |
| [6] |
J. Nicolas, S. Mura, D. Brambilla, N. Mackiewicz, P. Couvreur, Des. Chem. Soc. Rev. 42 (2013) 1147-1235. DOI:10.1039/C2CS35265F |
| [7] |
S. Swaminathan, J. Garcia-Amoros, A. Fraix, et al., Chem. Soc. Rev. 43 (2014) 4167-4178. DOI:10.1039/C3CS60324E |
| [8] |
K. Kataoka, A. Harada, Y. Nagasaki, Adv. Drug Deliv. Rev. 64 (2012) 37-48. DOI:10.1016/j.addr.2012.09.013 |
| [9] |
T. Terashima, T. Sugita, M. Sawamoto, Polym. J. 47 (2015) 667-677. DOI:10.1038/pj.2015.54 |
| [10] |
M. Matsumoto, T. Terashima, K. Matsumoto, M. Takenaka, M. Sawamoto, J. Am. Chem. Soc. 139 (2017) 7164-7167. DOI:10.1021/jacs.7b03152 |
| [11] |
Y. Koda, T. Terashima, M. Sawamoto, Macromolecules 49 (2016) 4534-4543. DOI:10.1021/acs.macromol.6b00998 |
| [12] |
J.H. Ko, A. Bhattacharya, T. Terashima, M. Sawamoto, H.D. Maynard, Polym. Chem. 57 (2019) 352-359. DOI:10.1002/pola.29187 |
| [13] |
G. Hattori, M. Takenaka, M. Sawamoto, T. Terashima, J. Am. Chem. Soc. 140 (2018) 8376-8379. DOI:10.1021/jacs.8b03838 |
| [14] |
A. Martien, S. Cohen, T. Wilhelm, et al., Nat. Mater. 9 (2010) 101-113. DOI:10.1038/nmat2614 |
| [15] |
C.L. Liu, C.H. Lin, C.C. Kuo, S.T. Lin, W.C. Chen, Prog. Polym. Sci. 36 (2011) 603-637. DOI:10.1016/j.progpolymsci.2010.07.008 |
| [16] |
Y.C. Lee, Y.S. Sun, J.Y. Liou, W.T. Chuang, Polymer 53 (2012) 5972-5981. DOI:10.1016/j.polymer.2012.10.031 |
| [17] |
L. Sun, W.M. Huang, Z. Ding, et al., Mater. Des. 33 (2012) 577-640. DOI:10.1016/j.matdes.2011.04.065 |
| [18] |
Z.Q. Cao, G.J. Wang, Chem. Rec. 16 (2016) 1398-1435. DOI:10.1002/tcr.201500281 |
| [19] |
T.Y. Wang, H.M. Zhu, H.G. Xue, Materials 9 (2016) 478. DOI:10.3390/ma9060478 |
| [20] |
Y.W. Pei, A.B. Lowe, P.J. Roth, Macromol. Rapid Commun. 38 (2017) 1600528. DOI:10.1002/marc.201600528 |
| [21] |
R. Cheng, F. Meng, C. Deng, H.A. Klok, Z. Zhong, Biomaterials 34 (2013) 3647. DOI:10.1016/j.biomaterials.2013.01.084 |
| [22] |
M. Motornov, Y. Roiter, I. Tokarev, S. Minko, Prog. Polym. Sci. 35 (2010) 174. https://www.sciencedirect.com/science/article/pii/S0079670009001026
|
| [23] |
B. Yan, Y. Zhang, C. Wei, et al., Polym. Chem. 9 (2018) 904-911. DOI:10.1039/C7PY01908D |
| [24] |
N. Jommanee, C. Chanthad, K. Manokruang, Carbohyd. Polym. 198 (2018) 486-494. DOI:10.1016/j.carbpol.2018.06.099 |
| [25] |
Y. Zhang, Y. Xu, C. Wei, Mater. Today Chem. 4 (2017) 172-179. DOI:10.1016/j.mtchem.2017.03.004 |
| [26] |
L. Zhang, Y. Wang, X.B. Zhang, et al., ACSAppl.Mater.Interfaces 9 (2017) 3388-3399. DOI:10.1021/acsami.6b14078 |
| [27] |
J.Y. Teo, B. Eng, W. Chin, et al., Nanomed. -Nanotechnol. 13 (2017) 431-442. DOI:10.1016/j.nano.2016.09.016 |
| [28] |
F.Y. Qiu, L. Yu, F.S. Du, Macromol. Rapid Commun. 38 (2017) 1700400-1700406. DOI:10.1002/marc.201700400 |
| [29] |
J. Feng, R.X. Zhuo, X.Z. Zhang, Prog. Polym. Sci. 37 (2012) 211-236. DOI:10.1016/j.progpolymsci.2011.07.008 |
| [30] |
H.F. Wang, H.Z. Jia, Y.F. Chu, et al., Macromol. Biosci. 14 (2014) 526-536. DOI:10.1002/mabi.201300414 |
| [31] |
W. Su, H.F. Wang, J. Feng, et al., J. Mater. Chem. 21 (2011) 6327.
|
| [32] |
X.L. Yang, X. Xing, J. Li, et al., RSC Adv. 9 (2019) 6003.
|
| [33] |
M.N. Ganivada, P. Kumar, P. Kanjilal, et al., Polym. Chem. 7 (2016) 4237-4245. DOI:10.1039/C6PY00615A |
| [34] |
C. Wei, Y. Zhang, H. Xu, et al., J. Mater. Chem. B 4 (2016) 5059.
|
| [35] |
H.M. Li, H. Jiang, M.N. Zhao, Y. Fu, X. Sun, Polym. Chem. 6 (2015) 1952-1960. DOI:10.1039/C4PY01623H |
| [36] |
H.F. Wang, H.Z. Jia, Y.F. Chu, et al., Macromol. Biosci. 14 (2014) 526-536. DOI:10.1002/mabi.201300414 |
| [37] |
V.T. Huynh, P. de Souza, M.H. Stenze, Macromolecules 44 (2011) 7888-7900. DOI:10.1021/ma2016503 |