 2019, Vol. 30
2019, Vol. 30 


b State Key Laboratory for Modification of Chemical Fibers and Polymer Materials, College of Materials Science and Engineering, Donghua University, Shanghai 201620, China;
c School of Materials Science and Energy Engineering, Foshan University, Foshan 528000, China
As typical oxide semiconductors, Fe/Co/Ni oxides have been widely explored in various fields, for example, nickel oxide (NiO) has turned out to be effective for applications in gas sensors, magnetic devices, catalysis, supercapacitors, and lithium-ion batteries, owing to its unique magnetic, optical, and electrical properties [1-4]. NiO is one of the earliest established gas sensing metal oxides due to its high sensitivity, good thermal stability and low cost [5-7]. The gas sensing performances depend greatly on the surface areas and nanostructures of the materials, as nanoporous structures could provide a large amount of adsorption sites and facilitate the diffusion and transfer of gas molecules. At present, porous NiO nanostructures with varieties of morphologies, such as nanosheets, nanospheres, nanoflowers, and hierarchical nanostructures, have been synthesized mainly via a hydrothermal or solvothermal process [8-12]. However, these methods usually lead to an ill-defined porous structure and uncontrollable morphology and porosity.
Ordered mesoporous metal oxides have attracted much interest owing to their ordered nanochannels, high specific surface areas, large accessible mesopores and semiconducting frameworks [13-18]. Compared to silica or carbon mesostructures with amorphous framework, the preparation of mesoporous metal oxides, especially mesoporous late transition metal oxides (e.g., Fe2O3, Co3O4, NiO) is more challenging [19-27]. Although ordered mesoporous NiO has been reported by hard-template method using mesoporous silica (SBA-15, KIT-6, etc.) as sacrificial template [28-31]. The hard template method involves tedious and complicated synthesis procedure, and thus is not suitable for the flexible and controllable synthesis of mesoporous materials with tunable pore sizes and structures. It is not applicable in mass production for practical applications. Much efforts based on soft-template method by using small surfactants like cetyltrimethy-lammonium bromide (CTAB) as templates has been also reported; however, the obtained porous NiO materials suffer from small pore size, amorphous framework or ill-defined mesostructures, because of the small hydrophobic size of the micelles and the relatively low crystallization temperature (< 350 ℃) of NiO [32-34]. To date, the synthesis of ordered mesoporous nickel oxides with large and well-interconnected ordered mesopores and highly crystalline walls through soft-template method still remains a great challenge.
The gas sensing performances of sensors based on metal oxides also depends on its structure and composition of the framework. To enhance the sensitivity of sensors based on p-type metal oxides, acceptor doping method is usually used to decrease the charge carrier concentrations of semiconductors, thus making p-type oxide semiconductor sensors more sensitive to analyte gases [35-37]. For example, Lee et al. reported that doping nanostructured CuO with Cr can significantly increase their sensing responses to NO2, due to the increase of resistance of the sensor caused by the Cr3+ ions in the crystalline CuO [35].
Recent studies have indicated that the co-assembly of organic and inorganic species to form ordered mesostructured materials can be well controlled by introducing chelating agents such as acetylacetone [38-41]. Inspired by these reports, we herein developed a facile solvent evaporation-induced co-assembly (EICA) method to synthesize mesoporous NiO, by using amphiphilic diblock copolymer polystyrene-b-poly(4-vinylpyridine) (PS-b-P4VP) as organic template and nickel acetylacetonate (Ni(acac)2) as the inorganic precursor. The obtained mesoporous NiO materials possess large main mesopores (~33.0 nm) and smaller secondary mesopores (4.0–10.0 nm) in the pore wall, high specific areas (~82.5 m2/g), and NiO nanocrystal-assembled walls. Furthermore, following the versatile EICA method, mesoporous Fe-doped NiO materials were obtained by introducing iron acetylacetonate (Fe(acac)3) source in the synthesis system. The obtained mesoporous Fe-doped NiO with similar porous structure and high surface area showed superior ethanol sensing performances of high sensitivity (9.5 ppm vs. 200 ppm), fast response-recovery speed (21 s/5 s) and good selectivity, owing to the high specific surface area and well-crystallized framework.
In a typical synthesis, 0.10 g of the block copolymer P4VP2k-b-PS10k (Mn = 12, 000 g/mol, PDI = 1.15) synthesized according to our previous work [42] was dissolved in 3 mL THF, forming solution A. 0.30 g of Ni(acac)2 was dissolved in 5 mL THF to form solution B. These two solutions were mixed to form a blue transparent colloidal solution. Then, the colloidal solution was poured into petri dishes to evaporate solvent at 25 ℃ for 12 h, followed by sequential heating at 40 ℃ for 12 h and solidification at 100 ℃ for another 24 h. Finally, the as- cast film was calcined at 350 ℃ for 3 h in N2 atmosphere (heating rate: 1 ℃/min) and 350 ℃ for 1 h in air (5 ℃/min). Similarly, mesoporous Fe-doped NiO materials were synthesized following the same method using 0.3 g of Ni(acac)2 and 0.012 g of Fe(acac)3 as the binary metal precursors.
Field-emission scanning electron microscopy (FESEM) images were obtained on a Zeiss Ultra 55 field-emission SEM (Germany) operated at 3 kV and 10 mA. Transmission electron microscopy (TEM) measurements were conducted on a JEOL JEM-2100F microscope operated at 200 kV. The samples for TEM measurements were suspended in ethanol and supported onto a carbon coated copper grid. Nitrogen sorption isotherms were measured with a Micromeritics Tristar 3020 analyzer at 77 K. Before measurements, all the samples were degassed in a vacuum at 180 ℃ for 8 h. The pore size distributions and the specific surface areas were calculated by using the Barrett-Joyner-Halenda (BJH) and Brunauer–Emmett–Teller (BET) method, respectively. The total pore volume (Vtotal) was calculated from the adsorbed amount at P/P0 = 0.995. Powder X-ray diffraction (PXRD) patterns were tested on a Bruker D4 X-ray diffractometer (40 kV, 40 mA) with Nifiltered Cu Kα radiation. Small-angle X-ray scattering patterns (SAXS) were obtained on a Bruker Nanostar U small-angle X-ray scattering system (40 kV, 35 mA) using Cu Kα radiation. Dynamic light scattering (DLS) were measured on Malvern Zetasizer ZS-90 analyzer.
Gas sensing measurements were tested on a gas sensing system of HW-30 A (Hanwei Electronics Co., Ltd., China) (Fig. S13 in Supporting information) using a gas sensor of side-heated type (Fig. S14a in Supporting information). The gas response of the gas sensor in this work was defined as S = Ra/Rg (for oxidizing gases) or S = Rg/Ra (for reducing gases). The response-recovery times are expressed as the time taken for the sensor output to reach 90% of its saturation after applying or switching off the gas in a step function. More details are in the Electronic Support Information.
The synthetic process of mesoporous NiO materials is illustrated in Scheme 1. In this work, the amphiphilic diblock copolymer PS-b-P4VP was chosen as organic template and the metal complex Ni(acac)2 as nickel source. THF was used as the solvent because it is a good solvent for both of the PS-b-P4VP and Ni(acac)2. Upon mixing, they formed spherical micelles in the solution, due to the strong coordination interactions between the N atoms of the hydrophilic P4VP block and the Ni atoms. After the evaporation of the THF, the uniform micelles gradually aggregate and assemble to form a hybrid organic-inorganic nanocomposite film. The film was then annealed in N2 atmosphere and air respectively to decompose the organic template and crystallize the framework, and mesoporous NiO materials with large pores and nanocrystal- assembled walls were obtained.

|
Download:
|
| Scheme 1. The synthetic process of the ordered mesoporous NiO. | |
{kind=link}
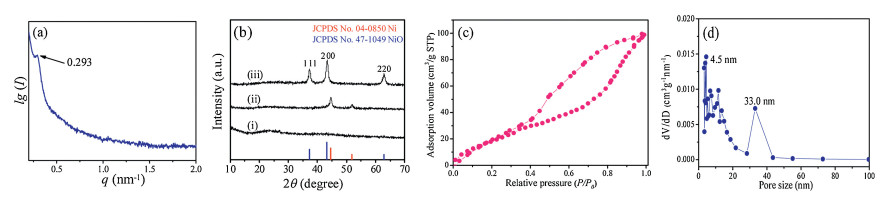
When mixing the PS-b-P4VP/THF and the Ni(acac)2/THF solutions, a green transparent colloidal solution with an obvious Tyndall Effect was immediately formed (Fig. S1 in Supporting information). It indicates that PS-b-P4VP block copolymers can interact with Ni(acac)2 directly and form colloidal hybrid micelles in THF. It is worth mentioning that, this phenomenon could be attributed to the strong nitrogen-metal coordination interactions (N→Ni) between the P4VP segments and nickel species in the solution. The dynamic light scattering (DLS) measurement shows a narrow size distribution of about 21.0 nm (Fig. S2 in Supporting information), confirming the formation of PS-b-P4VP/Ni(acac)2 spherical micelles with the hydrophobic PS block as a core and the P4VP/Ni(acac)2 as a shell. When the colloidal solution was cast on a glass Petri dish for evaporation of THF and further annealed at 100 ℃, and a homogeneous organic/inorganic hybrid film was formed. The small angle X-ray scattering (SAXS) patterns of the hybrid film displayed a well-resolved peak at q value of 0.293 nm-1 (Fig. 1a), and the corresponding d-spacing was calculated to be 21.5 nm (d100 = 2π/q). To obtain mesoporous NiO with well crystallized walls while maintaining the ordered mesoporous nanostructure, a template-carbonization supporting strategy was employed during the calcination. Firstly, the hybrid film was annealed at 350 ℃ in N2 atmosphere, during which the Ni(acac)2 was decomposed into Ni (JCPDS No. 04-0850) nanoparticles according to X-ray diffraction measurement (Fig. 1b, curve ⅱ), while the PS block was transformed into a rigid residual carbon to support the mesostructure, and the obtained black mNi/C sample was further calcined at 350 ℃ in air to remove the supporting carbon and simultaneously induce crystallization of nickel oxide, leading to mesoporous NiO with cubic phase mesoporous NiO (JCPDS No. 47-1049) (Fig. 1b, cuver ⅲ). The nitrogen adsorption-desorption isotherms of the mNiO-350-air sample show a typical Ⅳ-type hysteresis loop (Fig. 1c), indicating a well-defined mesostructure with large pores, and the specific surface area and pore volume are calculated to be 82.5 m2/g and 0.16 cm3/g, respectively. The obtained materials have dual pore size distributions centered at 33.0 nm and 4.5 nm, attributed to the main mesopores formed by decomposition of PS-b-P4VP and the interstitial voids of the packed NiO nanocrystals in the wall, respectively (Fig. 1d).

|
Download:
|
| Fig. 1. (a) SAXS pattern of the PS-b-P4VP/Ni(acac)2 nanocomposite. (b) XRD patterns of the (ⅰ) PS-b-P4VP/Ni(acac)2 nanocomposite, (ⅱ) mNi/C-350-N2 and (ⅲ) mNiO-350-air samples. (c) The nitrogen adsorption-desorption isotherms. (d) The corresponding pore-size distribution curves of the mNiO-350-air. | |
{kind=link}
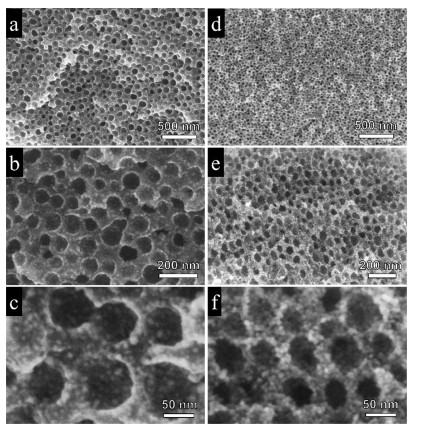
SEM observation indicates a short-range ordering of mesopores in the mNi/C-350- N2 (Figs. 2a–c).and mNiO-350-air samples (Figs. 2d and e). The mNi/C-350-N2 obtained after calcination in N2 possess an ultralarge mesopores of 45.0 nm with small Ni nanoparticles (~4.5 nm) uniformly distributed in the carbon matrix, which was derived from the pyrolysis of Ni(acac)2 (Fig. S3 in Supporting information). The enlargement of mesopore size is mainly due to the pore expansion during the pyrolysis of nickel acetylacetonate in inert atmosphere, because it can generate a large amount of gaseous mixture of carbon monoxide, carbon dioxide and water that expands the mesopores. The Raman spectra of mNi/C-350-N2 sample showed a distinct pair of broad bands at 1340 cm-1 and 1583 cm-1, which could be attributed to amorphous carbon and graphitic carbon structures respectively, indicating the existence of residual carbon (Fig. S4 in Supporting information). After calcination in air to remove the carbon residue completely, the ordered mesostructure was well maintained despite of the framework shrinkage by calcination. As estimated from the SEM image, the average pore size of mNiO-350-air decreased to about 36.0 nm, and the grain size of NiO nanoparticles in the pore wall slightly increased to about 6.0 nm after calcination in air. It is worth mentioning that the mNiO-350-air materials possess a unique porous structure with dual mesopores distribution. As shown in Fig. 2f, different from conventional mesoporous early-transition metal oxides (e.g., TiO2, WO3) with relatively dense pore walls synthesized by soft-templating method [39-41], the pore walls of the mNiO-350-air were found to be composed of uniform randomly-packed NiO nanocrystals. This hierarchical nanostructure can provide high surface areas and facilitate the diffusion and transport of guest molecules (e.g., gaseous molecules) within the porous materials, making them good candidates for applications in heterogeneous catalysis and gas sensing.

|
Download:
|
| Fig. 2. SEM images of the ordered mesoporous Ni/C (a–c) and ordered mesoporous NiO (d–f) obtained after calcination at 350 ℃ in N2 and 350 ℃ in air, respectively. | |
{kind=link}
The optimal synthesis condition was achieved by tailoring the Ni(acac)2/P4VP-b-PS mass ratio and annealing process. It was found that the ideal mesostructure was obtained at the ratio of 3.0 (Fig. S6 in Supporting information), and the exorbitant concentration and too lower concentration of Ni(acac)2 are unfavorable to forming spherical micelles via co-assembly. The annealing temperature is also an important factor, the well-defined mesostructure could not be maintained at temperatures higher than 400 ℃, and the mesostructure completely collapsed after annealing at 450 ℃ in air (Fig. S7 in Supporting information). The framework is brittle because the pore wall is built up by the non-closely packed NiO nanoparticles. Therefore, a precise control over annealing process below 400 ℃ is needed to prevent collapse of mesostructure by slowing down the crystallization of NiO and the combustion speed of residual carbon in the pore wall.
The choice of the block copolymer and the nickel source is also vital to ensure the successful synthesis of mesoporous NiO (Fig. S8 in Supporting information). The appropriate coordination interaction between the P4VP block and Ni(acac)2, and the organic ligand (-acac) guarantees the formation of small NiO nanocrystals. When PS-b-P4VP was replaced with PEO-b-PS, a transparent solution without Tyndall effect was obtained which indicates no hybrid micelles was formed (Fig. S8a), implying the absence of strong interaction between the PEO block and Ni(acac)2. As a result, the NiO-350-N2 sample exhibited a nonporous structure (Fig. S8d). Futhermore, when Ni(acac)2 was replaced with inorganic nickel salts like Ni(NO3)2·6H2O and NiCl2·6H2O, a cloudy pecusor solution was observed during synthesis (Fig. S8a). This may be expalined as follows. Compared to the neutral system of the Ni(acac)2/THF solution, the weak acidity of the Ni(NO3)2/THF and NiCl2/THF solutions can protonize the P4VP block, forming organic salts that can not be well dissolved in the THF based solutions, not favoring for a successful co-assembly process. Not surprisingly, the bulk materials composed of large crystals instead of mepoporous NiO was obtained in these cases (Figs. S8b and c).
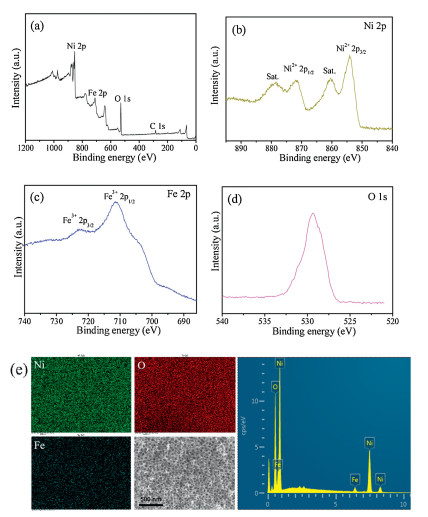
It is worth mention that, this facile method can be extended to synthesis of other mesoporous late-transition metal oxidesby using metalacetylacetone complexes asinorganic precusors. Forexample, following the same method, mesoporous Fe2O3 and Co3O4 can be obtained by using Fe(acac)3 and Co(acac)3 as the inorganic precursor, respectively (Fig. S9 in Supporting information). Importantly, by using Ni(acac)2 and Fe(acac)3 as binary metal precursor, a series of mesoporous Fe-doped NiO materials can be obtained following the same synthesis procedure (Fig. S10 in Supporting information). Taking the mesoporous Fe-doped NiO (Fe content: 3 at%) for example, the sample maintains a dual mesoporosity with uniform large main pores of ~32.5 nm and smaller pores of 4.0–11.5 nm, high specific surface areas (~74.8 m2/g) and large pore value (~0.167 cm3/g) (Figs. S10 and S11 in Supporting information). The XRD pattern of 3 at% Fe-NiO shows nearly the same diffraction peaks as the pure NiO sample (Fig. S12 in Supporting information), indicating the atomic doping of Fe into NiO matrix. The X-ray photoelectron spectroscopy (XPS) measurement shows the core level peaks of Ni 2P, O 1s and Fe 2p (Figs. 3a–d), and the peaks at 723.1 eV (Fe3+ 2p1/2) and 711.6 eV (Fe3+ 2p3/2), confirming the incorporation of Fe3+ into Ni2+ of the NiO crystal lattice. The energy dispersive spectrometer (EDS) spectrum further confirm the existence of Fe element, and the elemental mapping show that the three elements (Ni, Fe, O) were uniformly distributed in the mesoporous Fe-NiO materials (Fig. 3e).

|
Download:
|
| Fig. 3. X-ray photoelectron spectroscopy (XPS) measurements of the 3 at% Fe-doped NiO. (a) Wide-survey XPS, (b) Ni 2p spectra, (c) Fe 2p spectra, (d) O 1s spectra, (e) Elemental mapping (Ni, Fe, O) and EDS spectrum of the mesoporous 3 at% Fe-doped NiO. | |
{kind=link}
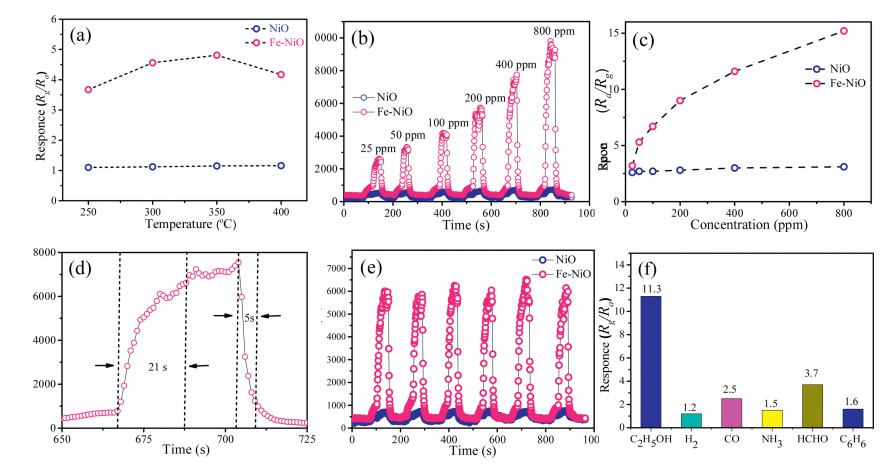
NiO is a typical p-type semiconductor which shows great potentials in gas sensing applications. In this study, the mesoporous NiO-based sensor was fabricated for studying their gas sensing properties toward gaseous ethanol. As a reducing gas, when the C2H5OH molecules was adsorbed on the surface of NiO, it would be oxidized by the pre-adsorbed oxygen species (O-, O2-, O2-) at high temperatures, and the carrier (i.e., hole) concentration of the p-type NiO would decrease, resulting in a rise of the resistance signal. Upon exposure to air, ethanol molecules desorb from NiO surface, and the resistance would recover to its initial value. In order to obtain the optimal working temperature, the mesoporous NiO-based sensor was firstly tested towards 50 ppm ethanol at temperatures of 250–400 ℃. It was found that, the pure mesoporous NiO-based sensor shows no changes of sensing response to ethanol vapor at the working temperature of 250– 400 ℃, which means poor sensing performance. By contrast, in the case of the mesoporous Fe-doped mesoporous NiO sensor, the response continuously increased until the operating temperature reached 350 ℃ and then decreased with the increase of temperature. Therefore, the optimal working temperature of the sensor is around 350 ℃ (Fig. 4a), and the 3 at% Fe-NiO-based sensor showed highest response among mesoporous NiO samples with different Fe doping content (Fig. S15 in Supporting information). Then, the sensor was tested for 25–800 ppm ethanol at 350 ℃. As shown in Figs. 4b–d, with the increase of C2H5OH concentration, the response (Rg/Ra) increased from 3.5 to 15.1 for the mesoporous 3 at% Fe-doped mesoporous NiO sensor, while the pure mesoporous NiO sensor exhibit poor sensing performance. The response and recovery time of mesoporous Fe-doped NiO sensor was calculated to be 21 s and 5 s, respectively, indicating a fast response-recovery speed. As an important parameter for a gas sensor, the selectivity is also crucial for practical applications. In this study, five kinds of typical vaporous molecules, including hydrogen, carbon monoxide, ammonia, formaldehyde and benzene, were tested as interfering gases. As shown in Fig. 4f, the response value of the sensor to ethanol was at least three times higher than the five interfering gases. Furthermore, the obtained gas sensors show an excellent cycling performance in response to 200 ppm ethanol, (Fig. 4e and Fig. S16 in Supporting information), indicating a long-term stability of mesoporous Fe-doped NiO sensor and laying the foundation for practical applications.

|
Download:
|
| Fig. 4. (a) The response of mesoporous NiO–based sensor to 50 ppm ethanol at different temperatures. (b) The response-recovery curves of the mesoporous NiO-based sensor to ethanol at different concentrations (25–800 ppm) at 350 ℃. (c) The response of the sensor vs. ethanol concentrations. (d) The response-recovery curve of the mesoporous NiO-based sensor to 200 ppm of ethanol at 350 ℃. (d) Repeating response-recovery curves of the sensor to 200 ppm ethanol. (e) The responses of the mesoporous NiO-based sensor to different gases of 400 ppm at 350 ℃. | |
{kind=link}
The increase of the gas response induced by doping NiO with Fe could be explained by the change of carrier concentrations [36, 37, 43]. As a typical p-type semiconductor, the carrier concentration of NiO is dominated by number of vacancy (or hole). When Fe3+ was incorporated into Ni2+ sites, the hole concentration would rise according to the charge compensation mechanism (1) and (2), and it resulted in an increase of sensor resistance, which was confirmed by the sensing tests. Thus, it can be concluded that the higher vacancy concentration formed in air would lead to a smaller variation in density of charge carrier by the gas sensing reaction, and lower vacancy concentration would be more advantageous for achieving a high gas sensing response. As a result, when exposing to reducing gas, the sensor based on mesoporous Fe-doped NiO could show higher response owning to the higher variation of the charge carrier concentrations.
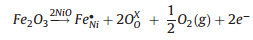
|
(1) |
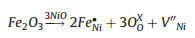
|
(2) |
In summary, a straightforward organic-inorganic co-assembly method has been develped for the controllable and flexible synthesis of mesoporous NiO and Fe-doped NiO materials with large pore size and nanocrystal-assembled walls by using amphipathicdiblock copolymerPS-b-P4VPasorganic templates andmetal complex Ni(acac)2 as inorganic source. The mesoporous NiO materials have large main mesopores (33.0 nm) and smaller secondary mesopores (4.0–10.0 nm) in the crystalline pore wall, high specific areas (82.5 m2/g), and NiO nanocrystal-assembled walls. Fe elements have been successfully employed to dope the NiO framework without damaging the porous structure in the range of 1–5 at% by using Ni(acac)2 and Fe(acac)3 as binary metal precursor. The obtained mesoporous Fe-doped NiO based gas sensors show superior sensing performances toward ethanol with high sensitivity, fast response-recovery speed (21 s/5 s) and good selectivity, which is due to their high specific surface area, dual mesoporosity and well-crystallized Fe-doped framework with high vacancy concentration. Based on the unique co-assembly of nitrogencontaining block copolymer and metal acetone complexes, various mesostructured transition metal oxide semiconductors with high surface area and well-defined porous structure can be controllably and flexibly for various applications such as catalysis, energy storage and conversion, chemical sensors, etc.
AcknowledgmentsThis work was supported by the NSF of China (Nos. 51372041, 51422202, 21673048, 21875044, 51822202 and 51772050), Key Basic Research Program of Science and Technology Commission of Shanghai Municipality (No. 17JC1400100), Youth Top-notch Talent Support Program of China, Shanghai Rising-Star Program (No. 18QA1400100), Youth Top-notch Talent Support Program of Shanghai, DHU distinguished Young Professor Program and the Fundamental Research Funds for the Central Universities.
Appendix A. Supplementary dataSupplementary material related to this article canbefound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.01.019.
| [1] |
Q. Dong, S. Yin, C.S. Guo, et al., Appl. Catal. B 147 (2014) 741-747. DOI:10.1016/j.apcatb.2013.10.007 |
| [2] |
Y.Q. Zhu, H.Z. Guo, Y. Wu, et al., J. Mater. Chem. A 2 (2014) 7904-7911. DOI:10.1039/c4ta00257a |
| [3] |
L.X. Liu, Y.Y. Guo, Y.P. Wang, et al., Electrochim. Acta 114 (2013) 42-47. DOI:10.1016/j.electacta.2013.09.152 |
| [4] |
D.P. Chen, X.L. Wang, Y. Du, et al., Cryst. Growth Des. 12 (2012) 2842-2849. DOI:10.1021/cg201676u |
| [5] |
P. Rai, J.W. Yoon, H.M. Jeong, et al., Nanoscale 6 (2014) 8292-8299. DOI:10.1039/C4NR01906G |
| [6] |
F. Li, Y. Chen, J. Ma, RSC Adv. 4 (2014) 14201-14205. DOI:10.1039/C4RA00182F |
| [7] |
H. Kim, J. Lee, Sens. Actuators B-Chem. 192 (2014) 607-627. DOI:10.1016/j.snb.2013.11.005 |
| [8] |
P. Justin, S.K. Meher, G.R. Rao, J. Phys. Chem. C 114 (2010) 5203-5210. DOI:10.1021/jp9097155 |
| [9] |
B. Cheng, Y. Le, W. Cai, J. Yu, J. Hazard. Mater. 185 (2011) 889-897. DOI:10.1016/j.jhazmat.2010.09.104 |
| [10] |
S.K. Meher, P. Justin, G.R. Rao, Electrochim. Acta 55 (2010) 8388-8396. DOI:10.1016/j.electacta.2010.07.042 |
| [11] |
C. Yuan, X. Zhang, L. Su, B. Gao, L. Shen, J. Mater. Chem. 19 (2009) 5772-5777. DOI:10.1039/b902221j |
| [12] |
M.W. Xu, S.J. Bao, H.L. Li, J. Solid State Electrochem. 11 (2007) 372-377. DOI:10.1007/s10008-006-0155-6 |
| [13] |
D. Gu, F. Schuth, Chem. Soc. Rev. 43 (2014) 313-344. DOI:10.1039/C3CS60155B |
| [14] |
Y. Ren, Z. Ma, P.G. Bruce, Chem. Soc. Rev. 41 (2012) 4909-4927. DOI:10.1039/c2cs35086f |
| [15] |
H. Kim, E. Lim, C. Jo, et al., ACS Nano 8 (2014) 8968-8978. DOI:10.1021/nn501972w |
| [16] |
C. Jo, J. Hwang, H. Song, et al., Adv. Funct. Mater. 23 (2013) 3747-3754. DOI:10.1002/adfm.201202682 |
| [17] |
B. Roose, K.C. Gödel, S. Pathak, et al., Adv. Energy Mater. 6 (2016) 1501868. |
| [18] |
X. Wang, L. Bai, H. Liu, et al., Adv. Funct. Mater. 28 (2018) 1704208. DOI:10.1002/adfm.201704208 |
| [19] |
J. Lee, O.M. Christopher, S.C. Warren, et al., Nat. Mater. 7 (2008) 222-228. DOI:10.1038/nmat2111 |
| [20] |
P.D. Yang, D.Y. Zhao, D.I. Margolese, et al., Chem. Mater. 11 (1999) 2813-2826. DOI:10.1021/cm990185c |
| [21] |
P.D. Yang, D.Y. Zhao, D.I. Margolese, et al., Nature 396 (1998) 152-155. DOI:10.1038/24132 |
| [22] |
P. Zhang, H. Lu, Y. Zhou, et al., Nat. Commun. 6 (2015) 8446. DOI:10.1038/ncomms9446 |
| [23] |
A.S. Poyraz, C.H. Kuo, S. Biswas, et al., Nat. Commun. 4 (2013) 2952. DOI:10.1038/ncomms3952 |
| [24] |
F.X. Ma, H. Hu, H.B. Wu, et al., Adv. Mater. 27 (2015) 4097-4101. DOI:10.1002/adma.201501130 |
| [25] |
L. Zhang, H.B. Wu, X.W.D. Lou, Adv. Energy Mater. 4 (2014) 1300958. DOI:10.1002/aenm.201300958 |
| [26] |
L. Han, D. Xu, Y. Liu, et al., Chem. Mater. 26 (2014) 7020-7028. DOI:10.1021/cm5033139 |
| [27] |
H. Li, Y. Liu, X. Cao, et al., Angew. Chem. Int. Ed. 56 (2017) 806-811. DOI:10.1002/anie.201611012 |
| [28] |
X. Lai, G. Shen, P. Xue, et al., Nanoscale 7 (2015) 4005-4012. DOI:10.1039/C4NR05772D |
| [29] |
H. Liu, G.X. Wang, J. Liu, S.Z. Qiao, H. Ahnc, J. Mater. Chem. 21 (2011) 3046-3052. DOI:10.1039/c0jm03132a |
| [30] |
F. Jiao, A.H. Hill, A. Harrison, J. Am. Chem. Soc. 130 (2008) 5262-5266. DOI:10.1021/ja710849r |
| [31] |
Y.G. Wang, Y.Y. Xia, Electrochim. Acta 51 (2006) 3223-3227. DOI:10.1016/j.electacta.2005.09.013 |
| [32] |
D.D. Zhao, M.W. Xu, W.J. Zhou, et al., Electrochim. Acta 53 (2008) 2699-2705. DOI:10.1016/j.electacta.2007.07.053 |
| [33] |
S. Banerjee, A. Santhanam, A. Dhathathreyan, P.M. Rao, Langmuir 19 (2003) 5522-5525. DOI:10.1021/la034420o |
| [34] |
P.A. Nelson, J.M. Elliott, G.S. Attard, J.R. Owen, Chem. Mater. 14 (2002) 524-529. |
| [35] |
K.M. Kim, H.M. Jeong, H.R. Kim, et al., Sensors 12 (2012) 8013-8025. DOI:10.3390/s120608013 |
| [36] |
J.K. Yoon, H.J. Kim, I.D. Kim, J.H. Lee, Nanotechnology 24 (2013) 444005. DOI:10.1088/0957-4484/24/44/444005 |
| [37] |
H.J. Kim, K.I. Choi, K.M. Kim, et al., Sens. Actuators B-Chem. 171 (2012) 1029-1037. |
| [38] |
Z. Sun, B. Sun, M. Qiao, et al., J. Am. Chem. Soc. 134 (2012) 17653-17660. DOI:10.1021/ja306913x |
| [39] |
J. Zhang, Y. Deng, D. Gu, Adv. Energy Mater. 1 (2011) 241. DOI:10.1002/aenm.201000004 |
| [40] |
J. Wei, Y. Ren, W. Luo, Chem. Mater. 29 (2017) 2211. DOI:10.1021/acs.chemmater.6b05032 |
| [41] |
Y. Zhu, Y. Zhao, J. Ma, J. Am. Chem. Soc. 139 (2017) 10365-10373. DOI:10.1021/jacs.7b04221 |
| [42] |
Y. Zhang, Q. Yue, L. Yu, Adv. Mater. 30 (2018) 1800345. DOI:10.1002/adma.201800345 |
| [43] |
H.J. Kim, J.W. Yoon, K.I. Choi, Nanoscale 5 (2013) 7066-7073. DOI:10.1039/c3nr01281f |