 2019, Vol. 30
2019, Vol. 30 


Synthetic polymer materials have infiltrated into every corner of modern society ranging from consumer plastics used in our everyday life to advanced nanomaterials in medicines and renewable energies. Benefited by rapid development of polymerization techniques, including living anionic and cationic polymerizations, ring-opening metathesis polymerization, and controlled radical polymerizations (CRPs), syntheses of these polymer materials have reached unprecedented levels to precisely control the polymers' compositions, molecular weights, chain structures, chain-sequence distributions and chain-end functionalities.
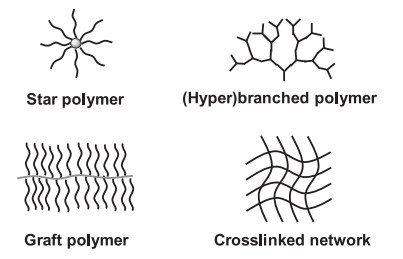
Among all these aspects of control, incorporating branched units into polymer chains becomes a popular and fast-growing area, i.e., polymer architecture. As compared to linear polymers that are constructed from the polymerization of difunctional monomers and contain two chain-end groups, polymers with branched structures possess at least one multifunctional monomer unit with functionality f > 2 and multiple chain-terminal groups. Thus, polymers with branched structures enjoy different features and properties, including higher solubility, lower solution/melt viscosity, less deformability and more chain-end functionality [1-3], as compared to their linear polymer counterparts. These differences provide polymers with branched structures intriguing applications as specialty adhesives, lubricants, drug delivery carriers, coating additives, and sensors. Although literature has reported many types of polymers in branched structures, such as star polymers, graft polymers, randomly branched polymers, hyperbranched polymers, dendrimers and polymer networks (Scheme 1), their structural differences are all related to the number, the functionality and the spatial placement of branching units in the polymer chains. For example, an ideal star polymer contains an n-functional branching point at the central core and n emanating arms. Graft polymers contain many 3- or 4-functional branching points, placed along a linear backbone with consequently hundreds of side chains [4, 5]. In randomly branched polymers, the branching units are randomly distributed throughout the macromolecule, similar to the structure of an insoluble gel, although the latter is a macroscopic network with "infinite" molecular weight. In addition, all these branching units could be introduced into polymers during the synthesis via using multifunctional initiators, multifunctional chain-transfer agents, multifunctional coupling agents, multivinyl crosslinkers, polymerizable initiators and/or polymerizable transfer agents [6].

|
Download:
|
| Scheme 1. Illustration of polymers with various branched structures: star polymer, graft polymer, (hyper)branched polymer and crosslinked polymer network. | |
{kind=link}
Polymerizations, as any chemical reactions, can be carried out in different media, including homogeneous systems (bulk or solution) or biphasic heterogeneous systems. For many polymerization techniques, the initial development always starts from the solution or bulk media since the one-phase homogeneous system is easy for setup to understand the reaction kinetics and mechanism. However, the biphasic heterogeneous systems, such as emulsion and suspension, provide unique features, including constant (or little changed) viscosity during the polymerization, discrete nanoreactors with compartmentalization effect and direct production of colloidal particles as the final products. Although biphasic heterogeneous systems by definition include any mixture of two immiscible liquids, polymerizations in practice are primarily carried out in normal oil-in-water (O/W) system and inverse water-in-oil (W/O) system. In particular, polymerizations in the O/W system, i.e., aqueous dispersed media, are more popularly applied to produce waterborne latexes and hydrophobic polymers due to the hydrophobic nature of most petroleum-based vinyl monomers and water as the most green and inexpensive solvent. In contrast, the inverse W/O biphasic media are used to prepare hydrophilic nanogels, ultrahigh-molecular-weight polyacryamides and inorganic nanoparticles. Meanwhile, some intrinsic features of these biphasic heterogeneous systems, such as the use of protic solvents as one phase, also post requirements on suitable polymerization techniques. In practice, radical-based polymerization are particularly used in the biphasic heterogeneous systems, which includes the conventional radical polymerization (FRP) and the recently developed CRPs, also known as reversible deactivation radical polymerization (RDRP) techniques [7, 8].
This review is dedicated to celebrate my Master thesis advisor, Prof. Shoukuan Fu's 80th birthday by discussing my personal journey on using radical polymerization in aqueous dispersed media to produce polymers with various branched structures. As Prof. Fu is an internationally renowned polymer chemist by establishing his research expertise on polymerizations in various biphasic heterogeneous media, I carefully chose this topic by combining radical polymerizations in aqueous dispersed media and polymers with branched structures into one short article. The literature to be discussed focuses on several projects I and coworkers accomplished in the past 20 years at different stages of my career, including the synthesis of star polymers, graft polymers, hyperbranched polymers, polymeric nanogels at Fudan University, Carnegie Mellon University, the University of California at Berkeley and the University of Notre Dame. As Prof. Fu, an academic mentor and a personal friend of mine, has watched me closely in the past two decades as I went through each of these stops, I expect this review by summarizing several of my accomplished projects using polymerization in aqueous dispersed media will be a special gift for celebrating this important event.
2. My passage to polymerization in aqueous dispersed mediaBy the summer of 1998, I have finished my sophomore year at Fudan University and started to seek a research opportunity in our Department of Macromolecular Science. It was this moment when Dr. Yiqiang Zhao, then a master student in Prof. Fu's group and the resident assistant (i.e., rector) of our 9644 class, influenced me to join the Fu lab and start my first-ever research project: the use of microemulsion polymerization to produce colloidal hydrogel. This project gave me the opportunity to know Prof. Fu who gradually guided me into the world of polymerization in aqueous dispersed media, e.g., emulsion polymerization. At that time, Prof. Fu's group had about 20 members and was actively developing various polymer materials using different polymerizations in microemulsion, soap-free emulsion, miniemulsion, seeded emulsion, dispersion, suspension, inverse microemulsion, as well as polymerization from surface. Such diversified research projects naturally provided me an opportunity to learn different polymerization techniques and research tricks, all new to a 3rd-year undergraduate student. What fascinated me more was Prof. Fu's approachable personality and open working style that helped maintain a very constructive group environment as a big family to facilitate members exchanging ideas, discussing research problems and hanging out in the spare time. This research experience during my undergraduate study not only taught me knowledge on polymer chemistry that significantly influenced my future research projects, but also determined my selection of various institutes in my later academic development. Below, I will first briefly introduce the radical polymerization in various types of aqueous dispersed media before discussing the several projects I accomplished at various stages by using polymerization in aqueous dispersed media.
3. Radical polymerization in various aqueous dispersed mediaSeveral important normal-phase O/W aqueous dispersed systems are briefly introduced here. Radical polymerization of vinyl monomers in each system produces stable latex particles or polymer beads that could be directly cast into films or be purified by removing the surfactant/stabilizers to recover the linear or branched polymers soluble in organic solvents. Based on the size of latex particles and the nucleation mechanism during the polymerization, these radical polymerizations in aqueous dispersed media (Scheme 2) can be classified as suspension polymerization, emulsion polymerization, miniemulsion polymerization, microemulsion polymerization and solution/precipitation polymerization (or dispersion polymerization).

|
Download:
|
| Scheme 2. Polymerizations in various biphasic aqueous dispersed media. | |
{kind=link}
Suspension polymerization [9] is kinetically similar to bulk or solution polymerization as the polymerization only proceeds in the dispersed droplets. These discrete reaction loci in the range of 10 mm to 1 mm are dispersed in the continuous phase, e.g., water, via vigorous agitation and small amount of stabilizers added to hinder coalescence of droplets. Typical polymers produced by aqueous suspension polymerization include polystyrene, poly (vinyl chloride), polyacrylates, poly(vinyl acetate) and their copolymers.
Emulsion polymerization [10-12] is widely used in industries to synthesize large quantities of latexes for applications in coatings, paints, adhesives as well as bulk polymers. Before polymerization, an emulsion is composed of monomer-solubilized micelles (ca. 5– 10 nm), monomer droplets (within the size 1–10 mm or larger) and the monomer-saturated continuous phase, such as water. A typical emulsion polymerization contains three kinetic intervals: 1) particle nucleation period that ends by the mark of the disappearance of micelles; 2) propagation period that has coexisted polymerizing latexes and monomer droplets and ends by the mark of the disappearance of monomer droplets; 3) monomer depletion period that reacts the monomer inside each polymerizing latex until the end of polymerization. Many theoretical descriptions concerning the unique kinetics of emulsion polymerization have been developed over the years [13]. The most commonly recognized kinetic theory was established by Harkins [14], Smith and Ewart [15]. Surfactant is a key formulation component used in emulsion polymerization and it could be categorized based on its charge nature in anionic, cationic, nonionic and zwitterionic surfactants. Meanwhile, an empirical HLB (hydrophile-lipophile balance) value of each surfactant could be determined as a means for predicting the surfactant's emulsification efficiency [16], in which a high HLB value often indicates superior emulsification capacity. As a special type of surfactant, reactive surfactants [17] (i.e., surfactants that are able to react during the emulsion polymerization process) have been applied in emulsion polymerization as they can be covalently bound into the macromolecular chains and ultimately improve the polymer film stability. Emulsion polymerization in general produces polymer latexes in size of 60–500 nm.
Miniemulsion polymerization [18-21] intends to eliminate the monomer mass transfer between discrete droplets through continuous media during the polymerization. Therefore, each monomer droplet is expected to go through nucleation during polymerization and considered as a "mini-bulk" reactor. Meanwhile, miniemulsion polymerization aims to produce submicronsized particles (< 500 nm) by using surfactant and more importantly ultrasonic homogenization to break monomer droplets and reduce Ostwald ripening (transfer of monomer from small droplets to large droplets to reduce the total surface energy of the system) so as to reach a metastable heterogeneous state. Although miniemulsion shares many similarities as emulsion polymerization, their distinct lies in the aspects of particle nucleation and mass transportation. No micelles or micron-sized monomer droplets are present in the miniemulsion [22] as it after sonication only contains submicron monomer droplets in relative stability. Very often, oil-soluble initiators are mixed with monomers before sonication so that the submicron droplets encapsulate all necessary components for the subsequent initiation and polymerization without any mass transfer through the continuous media. This feature provides miniemulsion and subsequent polymerization unique advantages to encapsulate guest molecules (inorganic or organic compounds) into each monomer droplet and produce hybrid nanomaterials. However, the generation of stable miniemulsion droplets requires high energy input during the sonication process, which limits its broad application in many scale-up productions in industries [23].
Microemulsion polymerization [24, 25] synthesizes polymersin a thermodynamically stable system and produces polymer nanoparticles with typical dimension in 10–50 nm. In the presence of large amount of surfactant, microemulsion could be formed spontaneously without any vigorous mixing of oil and water, although mild magnetic stirring is often used in lab setup to improve the kinetics of microemulsification. As compared to emulsion polymerization, microemulsion polymerization experiences two kinetic intervals instead of three due to the absence of monomer droplet from the starting [26]. In a modified microemulsion polymerization developed by Dr. Weihua Ming in Prof. Fu's group [27], continuous and slow addition of monomer to the polymerizing microemulsion system could dramatically increase the weight ratio of polymer to surfactant from < 10 wt% to about 25 wt% without disturbing the microemulsion stability. When the system was emulsified by anionic surfactant and initiated by redox initiators at ambient temperature, the polymerization could produce microemulsion latexes with particle size < 20 nm, polymer/surfactant weight ratio > 7:1 and polymeric microemulsion concentration in water as high as 40 wt% [28]. It was very interesting to note that further increasing the monomer/surfactant ratios above a critical level or using high initiator concentrations could trigger some microemulsion systems, suchaspoly(methylmethacrylate) (PMMA) and poly(ethyl methacrylate) (PEMA), to form transparent physical hydrogels via noncovalent latex interaction either during the polymerization or after a period of latex storage [29].
Dispersion polymerization [30, 31] as compared to the above aqueous dispersed media, is more frequently applied to systems using a dispersant phase, such as alcohol, other than water. Before polymerization, all monomers, initiators and other compounds are soluble in dispersant to form homogeneous solution. Initiation to polymerize the soluble monomers quickly produces oligomers that reach the critical length and precipitate to form biphasic heterogeneous system. In the presence of stabilizers, these precipitated polymers aggregate into small-nuclei assemblies surrounded by the stabilizers [32], which further absorb monomer, initiator and initiating radicals from the dispersant phase into the polymer particles for continuing growth and polymerization. Therefore, dispersion polymerization in principle is a solution/ precipitation polymerization, which is also similar to the polymerization-induced self-assembly (PISA) [33] although the PISA technique mainly focuses on the synthesis of block (segmented) copolymers in dispersed media. Dispersion polymerization is an attractive method for production of micron-size monodisperse polymer particles (1–15 mm) in a single batch process. The size and size distribution have demonstrated to be very much dependent on reaction conditions, including the nature of stabilizer, stabilizer concentration, monomer concentration and initiator concentration.
Compartmentalization effect in aqueous dispersed media refers to the physical confinement of reactants to a very small space [10]. As compared to polymerizations in bulk or solution, radical polymerizations in dispersed media often show different kinetics due to two specific results of compartmentalization, segregation effect and confinement effect [34]. The segregation effect refers to two species located in separate particles being unable to react, e.g., a decreased rate of bi-radical termination reaction; while the confinement effect refers to two species located in the same particle reacting at a higher rate because of their high concentration in a small particle. In terms of FRP, the consequence of compartmentalization effect is generally an increase of polymerization rate due to decreased bi-radical termination reaction. When CRP is used in biphasic dispersed systems, compartmentalization effects may play a more significant role on the polymerization kinetics and consequently on the polymer structures. As compared to FRP in aqueous dispersed media which only have compartmentalized propagating radicals species, the CRP systems in aqueous dispersed media involve more species, such as initiators, deactivators (nitroxides, Cu(Ⅱ)/ligand complexes, photocatalysts) and chain-transfer-agent fragments, in the compartmentalized space. To understand the effect of compartmentalization on the CRP kinetics and chain livingness, various simulation and modeling [35] have been carried out for nitroxide-mediated polymerization (NMP) [34, 36-38], atom transfer radical polymerization (ATRP) [39] and reversible addition fragmentation chain transfer (RAFT) polymerization [40, 41] in dispersed media. The results provide a comprehensive and sometimes complicated picture, in which the compartmentalization effect is dependent on a bracket of parameters, including the concentration and reactivity of deactivators, chain transfer agents, propagating radicals, particle sizes, as well as the partition coefficient of each species between the dispersed phase and water. In some cases, the distribution (or heterogeneity) of these species between different particles, such as monomer concentration variation [40], also shows influence on the polymerization kinetics.
4. Synthesis of crosslinked nanoparticles and nanocapsulesThe projects discussed in this section were accomplished during my M.S. study at Fudan University and my postdoctoral research at UC Berkeley. Although the motivations in these projects were different, their success all relied on the critical syntheses that utilized the techniques I learned in the Fu lab: polymerization in aqueous dispersed media, to produce polymeric particles containing crosslinked structures.
Immediately after joining Prof. Fu's group in the fall of 2000 as a M.S. student, I being inspired by several other graduate students in the group started to plan a project focusing on "preparation of a novel polymeric fluorescent nanoparticles" [42] using the modified microemulsion polymerization technique developed in the Fu group [27]. In this project, a crosslinked polystyrene nanoparticle with physically trapped pyrene molecules was synthesized via the modified microemulsion copolymerization of styrene, divinylbenzene and amino ethyl methacrylate hydrochloride (AEMH) as a hydrophilic monomer, in the presence of pyrene. After synthesis and purification, these crosslinked nanoparticles with hydrodynamic diameter Dh = 20–50 nm (Fig. 1A) showed high photoluminescence intensity, ca. 40 times higher than that of pyrene in toluene or styrene solution at the same concentration. The fluorescence intensity could be easily tuned based on the amount of pyrene molecules added, while the size of nanoparticles was affected by the use of AEMH monomer and the initiator 2, 20-azobis (2-amidinopropane)dihydrochloride (V50).

|
Download:
|
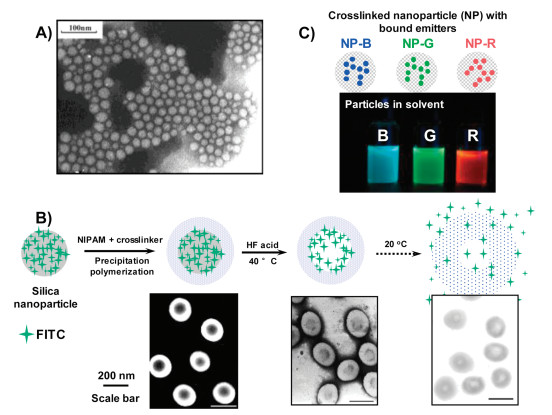
| Fig. 1. (A) Transmission electron microscopy (TEM) image of the fluorescent nanoparticles synthesized by the modified microemulsion polymerization. Reproduced with permission [42]. Copyright 2012, Springer. (B) Synthesis of PNIPAM crosslinked nanocapsules using sacrificial templates in precipitation polymerization. Reproduced with permission [45]. Copyright 2015, Elsevier. (C) Synthesis of light-emitting ink particles using miniemulsion polymerization. Reproduced with permission [47]. Copyright 2010, American Chemical Society. | |
{kind=link}
During thisproject, I started to explore the delivery and controlled release of guest molecules using crosslinked polymer capsules. For this purpose, I studied the possibility of using silica nanoparticles as sacrificial core templates in order to ultimately produce polymeric hollownanoparticles after removalof silica.During that time, several group members [43, 44] in the Fu group were also actively studying the synthesisofmulti-responsive nanoparticles andnanocontainers, which profoundly shaped my idea to accomplish this research. In 2005, we with several group members published our work in polymer "thermosensitive poly(N-isopropylacrylamide) nanocapsules with controlled permeability" [45]. In this project, thermosensitive polymer capsules based on crosslinked poly(N-isopropylacrylamide) (PNIPAM) were prepared in three steps (Fig. 1B): 1) synthesis of silica nanoparticles with surface-decorated isothiocyanate fluorescein (FITC) using a modified Stöber method [46], 2) precipitation polymerization of NIPAM and diacrylamide crosslinker around the silica nanoparticles to produce core-shell structured nanoparticles, 3) removal of silica core via HF acid etching to produce the targeted FITC-trapped PNIPAM nanocapsuleswith crosslinked structure.Characterization results clearly recorded the step-by-step construction of the core-shell particles and the final hollow capsules. The release kinetics of FITC from the capsule could be effectively affected by the environment, in which temperature above 32 ℃ dehydrated the PNIPAM shell and slowed down the release of FITC from the internal cavity.
The three-year M.S. study at Prof. Fu's group (2000–2003) gave me an opportunity to gain solid training on radical polymerization in aqueous dispersed media and be very familiar of the microemulsion, miniemulsion, emulsion and seeded emulsion polymerization. This piece of knowledge later not only powered my Ph.D. research at Carnegie Mellon University on using ATRP for synthesis of star polymers in miniemulsion [48] and hairy nanoparticles in seeded emulsion [49], but also salvaged my postdoc research at University of California (UC), Berkeley, where I developed polymer materials for cascade catalytic reactions and for dual light emission in organic light emitting diodes.
Since late 1990s, Prof. Fréchet has successfully used structurally defined dendrimers [50] and star polymers [51, 52] as catalyst carriers to achieve one-pot cascade reactions and free-energy driven catalytic pump [53], which vividly mimicked enzymes, the Nature's polymer catalysts, for being able to carrying competing reactions in crowded cellular milieu. However, the research was often limited by the complicated and inefficient synthesis of polymer carriers in both dendrimers and star polymers. As I joined the Fréchet group as a postdoc researcher in January 2009, my immediate goal was to develop a robust polymer carrier that had defined structure and modular reactive sitesforloading avarietyof catalytic species without repeated polymer synthesis and optimization. After several attempts ofothermethods, I quickly decided to use my skill on polymerization in aqueous dispersed media by designing a water-dispersible hairy nanoparticle, or core crosslinked star polymer, as a modular polymer catalyst carrier [54]. Specifically, the crosslinked core particle was synthesized via microemulsion copolymerization of structural monomer, crosslinker and azido-functionalized monomer, while the hair poly(ethylene glycol) PEG polymers were incorporated via coupling-onto route using efficient copper-catalyzed azide-alkyne cycloaddition (CuAAC) click reactions. This project demonstrated that the corenanoparticles with chemically inert composition could be functionalized at will by reacting with alkyne-containing "payloads", including organocatalysts, metal complexes and dyes, for quick access to a variety of polymer catalysts using one coupling protocolatmmol/gloading capacity.Thispieceofwork alsoinspired my future interest at the University of Notre Dame on developing hyperbranched polymers as unimolecular nanocontainers.
In a parallel project at UC Berkeley, my interest on designing a library of light emitting inks that could freely mix at various ratios to achieve dual emission drove me using miniemulsion polymerization to produce crosslinked light-emitting nanoparticles [47]. These nanoparticles each contained different Ru-complexes as red, green or blue emitters site-isolated in the crosslinked polymer nanoparticles. When dispersed in organic solvent, these functional nanoparticles behaved as R, G and B inks (Fig. 1C) that could be freely mixed and cast into smooth films for preparing lightemitting diodes with tunable electroluminescent colors.
5. Synthesis of hyperbranched polymers using microemulsion polymerizationAfter joining Notre Dame in 2011 to start my independent career, I targeted to solve a well-documented synthetic challenge by developing new methods for synthesis of structurally defined hyperbranched polymers in one pot. As the concept of one-pot synthesis of highly branched polymers was first reported in 1940s, the advantage and limitation in this one-pot polymerization of AB2 monomers was well discussed in literature and textbook. In sharp contrast from dendrimers that have elegant structure at the cost of sophisticated multi-step reactions [55-57], hyperbranched polymers feature with the facile one-pot polymerization, but suffer random bimolecular reactions with no control of polymer structures [58, 59].
The use of radical polymerization for synthesis of highly branched polymers experienced two stages of developments. Before the invention of CRP techniques, conventional radical copolymerization of divinyl crosslinkers in the use of high-concentration initiators or chain transfer agents represented an effective method to produce soluble branched polymers since the initiators and transfer agents could stop the propagation of one chain before it grew too long to reach macroscopic gelation [60, 61]. After CRP techniques were developed in the 1990s, they were rapidly applied in the synthesis of hyperbranched polymers via NMP [62, 63] and ATRP [64-66] of inimers (containing an initiator fragment and a vinyl group in one molecule), RAFT polymerization [67-71] of transmers (containing a chain transfer fragment and a vinyl group in one molecule) and copolymerization of crosslinkers using any of the three methods. However, all these reports prepared hyperbranched polymers in homogeneous media, in which the random bimolecular reactions, i.e., monomer-monomer, monomer-polymer and polymer-polymer reactions, in a continuous reaction media resulted in polymers with extremely broad molecular weight distribution (MWD) [62, 64, 66, 72].
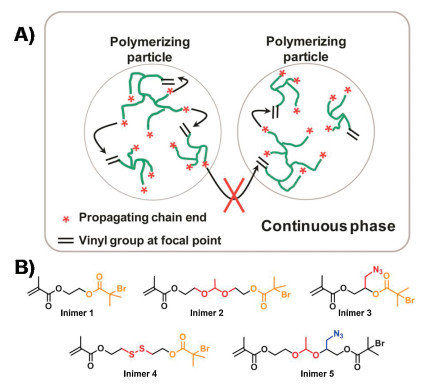
To improve the polymer structure without sacrificing the facile one-pot synthesis, I again turned my attention to the aqueous dispersed media. Instead of carrying out the polymerization in a homogeneous media, ATRP of an inimer in the microemulsion that used the micelles as confined space for segregating the random bimolecular reactions into each discrete nanoreactor successfully produced hyperbranched polymers with better-defined structure and low dispersity with Mw/Mn ~1.5 (Fig. 2A). By the end of polymerization, there was only one highly branched polymer per micelle and the molecular weight of polymer was directly determined by the micelle dimension. After the first publication in2012 [73], mygroup atNotreDamehas systematically investigated this method by exploring several experimental variables, including monomer species, Cu catalyst, ligands, varied amounts ofmonomers and surfactants. The new method exhibited great robustness in the polymerization of different inimers (Fig. 2B) and produced a series of hyperbranched polymers with varied compositions, degree of branching (DB = 0.26-0.41), molar mass (Mn = 103-106 g/mol), and low dispersity (Mw/Mn = 1.1–1.7) [74]. The obtained hyperbranched polymers exhibited superior loading efficiency of guest molecules as compared to traditional nanogels synthesized from the copolymerization of divinyl crosslinkers [75]. For instance, the hyperbranched polymerswith DB = 0.30 exhibited a 3-time higher loading efficiency of a molecular probe than the 5% crosslinked nanogel with similar dimension and reactive group density. Meanwhile, all hyperbranched polymers produced within the spatially confined micelles (i.e., latexes) contained hundreds of bromine dormant initiating sites. They could be subsequently used as macroinitiators for polymerization of another functional monomer to synthesize hyperstar polymers in core-shell structure. The composition and dimension of the core and shell segments could be independently controlled based on several experimental variables, including the monomer species, the feed ratios and the conversions. Very recently, this type of synthesis was further accomplished in a one-pot procedure via sequential polymerization of ABm monomer and a 2nd functional monomer in a O/W seeded emulsion [76]. The surfactant-protected emulsion latexes functioned as segregated reactors to effectively eliminate the undesired star-star coupling, achieving both high monomer conversion and fast polymerization at the same time.

|
Download:
|
| Fig. 2. (A) Conceptual illustration of polymerization of inimers in microemulsion using ATRP. Reproduced with permission [73]. Copyright 2012, American Chemical Society. (B) Structures of inimers applied in the microemulsion polymerization carrying varied compositions and functional groups. | |
{kind=link}
These hyperbranched polymers and hairy nanoparticles containing several defined domains within one nanostructure have been successfully applied as unimolecular nanocontainers to encapsulate and release active ingredients in nanomedicines, including ⅰ) co-delivery of two enzyme inhibitors to suppress the proliferation of triple-negative breast cancer cells [77] and ⅱ) protection and delivery of fragile siRNA molecules to target the amyotrophic lateral sclerosis (ALS) disease [78]. These drugpolymer conjugates could tolerate repeated lyophilization-redispersion without losing any drug activity and open opportunity to obtain drug-polymer conjugates in dispersible powder state for convenient storage and transportation.
6. Final remarksIt is my great honor to contribute this short review article in celebrating Prof. Shoukuan Fu's 80th birthday. Since joining Prof. Fu's group in 1998 at Fudan University as an undergraduate student, I have learned tremendously from Prof. Fu on not only the truth of research, but also the truth of life. In the past 20 years, Prof. Fu has watched me closely to develop my career, and to grow our family. The mentorship and friendship that Prof. Fu offers my family and myself will last long after this celebration event.
AcknowledgementThe authors thank the University of Notre Dame for financial support.
| [1] |
N. Hadjichristidis, H. Iatrou, M. Pitsikalis, J. Mays, Prog. Polym. Sci. 31 (2006) 1068-1132. DOI:10.1016/j.progpolymsci.2006.07.002 |
| [2] |
K. Matyjaszewski, Y. Gnanou, L. Leibler, Macromolecular Engineering: From Precise Macromolecular Synthesis to Macroscopic Materials Properties and Applications, Wiley-VCH, Weinheim, 2007.
|
| [3] |
Y.Y. Cui, Y.E. Ren, X.X. Liu, Chin. Chem. Lett. 23 (2012) 985-988. DOI:10.1016/j.cclet.2012.06.026 |
| [4] |
M. Zhang, A.H.E. Mueller, J. Polym. Sci. Part A:Polym. Chem. 43 (2005) 3461-3481. DOI:10.1002/pola.20900 |
| [5] |
H. Du, L.L. Gao, W.P. Zhu, Z.Q. Shen, Chin. Chem. Lett. 23 (2012) 879-882. DOI:10.1016/j.cclet.2012.05.022 |
| [6] |
C.J. Hawker, K.L. Wooley, Science 309 (2005) 1200-1205. DOI:10.1126/science.1109778 |
| [7] |
P.B. Zetterlund, S.C. Thickett, S. Perrier, E. Bourgeat-Lami, M. Lansalot, Chem. Rev. 115 (2015) 9745-9800. DOI:10.1021/cr500625k |
| [8] |
K. Min, K. Matyjaszewski, Cent. Eur. J. Chem. 7 (2009) 657-674. |
| [9] |
E. Vivaldo-Lima, P.E. Wood, A.E. Hamielec, A. Penlidis, Ind. Eng. Chem. Res. 36 (1997) 939-965. DOI:10.1021/ie960361g |
| [10] |
R.G. Gilbert, Emulsion Polymerization, Academic, San Diego, CA, 1995.
|
| [11] |
P.A. Lovell, Mohamed S. El-Aasser, Emulsion Polymerization and Emulsion Polymers, John Wiley & Sons, Inc., New York, 1997 p. 510.
|
| [12] |
C.S. Chern, Prog. Polym. Sci. 31 (2006) 443-486. DOI:10.1016/j.progpolymsci.2006.02.001 |
| [13] |
S.C. Thickett, R.G. Gilbert, Polymer 48 (2007) 6965-6991. DOI:10.1016/j.polymer.2007.09.031 |
| [14] |
W.D. Harkins, J. Am. Chem. Soc. 69 (1947) 1428-1444. DOI:10.1021/ja01198a053 |
| [15] |
W.V. Smith, R.H. Ewart, J. Chem. Phys. 16 (1948) 592-599. DOI:10.1063/1.1746951 |
| [16] |
W.C. Griffin, J. Soc. Cosmet. Chem. Jpn. 1 (1949) 311-326. |
| [17] |
A. Guyot, Adv. Colloid Interface Sci. 108- 109 (2004) 3-22. |
| [18] |
J.M. Asua, Prog. Polym. Sci. 27 (2002) 1283-1346. DOI:10.1016/S0079-6700(02)00010-2 |
| [19] |
K. Landfester, Macromol. Rapid Commun. 22 (2001) 896-936. DOI:10.1002/1521-3927(20010801)22:12<896::AID-MARC896>3.0.CO;2-R |
| [20] |
M. Antonietti, K. Landfester, Prog. Polym. Sci. 27 (2002) 689-757. DOI:10.1016/S0079-6700(01)00051-X |
| [21] |
J. Ugelstad, M.S. El-Aasser, J.W. Vanderhoff, J. Polym. Sci. Part C:Polym. Lett. 11 (1973) 503-513. DOI:10.1002/pol.1973.130110803 |
| [22] |
N. Bechthold, K. Landfester, Macromolecules 33 (2000) 4682-4689. DOI:10.1021/ma000061h |
| [23] |
J.M. Asua, Prog. Polym. Sci. 39 (2014) 1797-1826. DOI:10.1016/j.progpolymsci.2014.02.009 |
| [24] |
M. Antonietti, R. Basten, S. Lohmann, Macromol. Chem. Phys. 196 (1995) 441-466. DOI:10.1002/macp.1995.021960201 |
| [25] |
P.Y. Chow, L.M. Gan, Adv. Polym. Sci. 175 (2005) 257-298. |
| [26] |
F. Candau, Y.S. Leong, R.M. Fitch, J. Polym. Sci.:Polym. Chem. Ed. 23 (1985) 193-214. DOI:10.1002/pol.1985.170230120 |
| [27] |
W. Ming, F.N. Jones, S. Fu, Polym. Bull. (Berl.) 40 (1998) 749-756. DOI:10.1007/s002890050318 |
| [28] |
W. Ming, F.N. Jones, S. Fu, Macromol. Chem. Phys. 199 (1998) 1075-1079. DOI:10.1002/(SICI)1521-3935(19980601)199:6<1075::AID-MACP1075>3.0.CO;2-H |
| [29] |
W. Ming, Y. Zhao, J. Cui, S. Fu, F.N. Jones, Macromolecules 32 (1999) 528-530. DOI:10.1021/ma9813486 |
| [30] |
R. Arshady, Colloid Polym. Sci. 270 (1992) 717-732. DOI:10.1007/BF00776142 |
| [31] |
S. Kawaguchi, K. Ito, Adv. Polym. Sci. 175 (2005) 299-328. |
| [32] |
S. Shen, E.D. Sudol, M.S. El-Aasser, J. Polym. Sci. Part A:Polym. Chem. 32 (1994) 1087-1100. DOI:10.1002/pola.1994.080320611 |
| [33] |
N.J. Warren, S.P. Armes, J. Am. Chem. Soc. 136 (2014) 10174-10185. DOI:10.1021/ja502843f |
| [34] |
P.B. Zetterlund, M. Okubo, Macromolecules 39 (2006) 8959-8967. DOI:10.1021/ma060841b |
| [35] |
H. Tobita, F. Yanase, Macromol. Theory Sim. 16 (2007) 476-488. DOI:10.1002/mats.200700007 |
| [36] |
H. Meahata, C. Buragina, M. Cunningham, Macromolecules 40 (2007) 7126-7131. DOI:10.1021/ma070603w |
| [37] |
B. Charleux, Macromolecules 33 (2000) 5358-5365. DOI:10.1021/ma0002904 |
| [38] |
G. Delaittre, B. Charleux, Macromolecules 41 (2008) 2361-2367. DOI:10.1021/ma702498u |
| [39] |
Y. Kagawa, P.B. Zetterlund, H. Minami, M. Okubo, Macromol. Theory Sim. 15 (2006) 608-613. DOI:10.1002/mats.200600049 |
| [40] |
H. Tobita, Macromol. Theory Sim. 18 (2009) 108-119. DOI:10.1002/mats.200800069 |
| [41] |
H. Tobita, Polymers 3 (2011) 1944-1971. DOI:10.3390/polym3041944 |
| [42] |
H.F. Gao, Y.Q. Zhao, S.K. Fu, B. Li, M.Q. Li, Colloid Polym. Sci. 280 (2002) 653-660. DOI:10.1007/s00396-002-0670-7 |
| [43] |
L. Zha, Y. Zhang, W. Yang, S. Fu, Adv. Mater. 14 (2002) 1090-1092. DOI:10.1002/1521-4095(20020805)14:15<1090::AID-ADMA1090>3.0.CO;2-6 |
| [44] |
Y. Deng, W. Yang, C. Wang, S. Fu, Adv. Mater. 15 (2003) 1729-1732. DOI:10.1002/adma.200305459 |
| [45] |
H.F. Gao, W.L. Yang, K. Min, et al., Polymer 46 (2005) 1087-1093. DOI:10.1016/j.polymer.2004.11.078 |
| [46] |
W. Stöber, A. Fink, E. Bohn, J. Colloid Interface Sci. 26 (1968) 62-69. DOI:10.1016/0021-9797(68)90272-5 |
| [47] |
H. Gao, D.A. Poulsen, B. Ma, et al., Nano Lett. 10 (2010) 1440-1444. DOI:10.1021/nl100347p |
| [48] |
K. Min, H. Gao, K. Matyjaszewski, J. Am. Chem. Soc. 127 (2005) 3825-3830. DOI:10.1021/ja0429364 |
| [49] |
K. Min, H. Gao, J.A. Yoon, et al., Macromolecules 42 (2009) 1597-1603. DOI:10.1021/ma8026244 |
| [50] |
B. Helms, J.M.J. Fréchet, Adv. Synth. Catal. 348 (2006) 1125-1148. DOI:10.1002/adsc.200606095 |
| [51] |
B. Helms, S.J. Guillaudeu, Y. Xie, et al., Int. Ed. 44 (2005) 6384-6387. DOI:10.1002/anie.200502095 |
| [52] |
Y. Chi, S.T. Scroggins, J.M.J. Fréchet, J. Am. Chem. Soc. 130 (2008) 6322-6323. DOI:10.1021/ja8013456 |
| [53] |
M.E. Piotti, F. Rivera, R. Bond, C.J. Hawker, J.M. Fréchet, J. Am. Chem. Soc. 121 (1999) 9471-9472. DOI:10.1021/ja991879p |
| [54] |
V. Rodionov, H. Gao, S. Scroggins, et al., J. Am. Chem. Soc. 132 (2010) 2570-2572. DOI:10.1021/ja9104842 |
| [55] |
D.A. Tomalia, H. Baker, J. Dewald, et al., Polym. J. 17 (1985) 117-132. DOI:10.1295/polymj.17.117 |
| [56] |
A.W. Bosman, H.M. Janssen, E.W. Meijer, Chem. Rev. 99 (1999) 1665-1688. DOI:10.1021/cr970069y |
| [57] |
S.M. Grayson, J.M.J. Fréchet, Chem. Rev. 101 (2001) 3819-3867. DOI:10.1021/cr990116h |
| [58] |
Y.H. Kim, J. Polym. Sci. Part A:Polym. Chem. 36 (1998) 1685-1698. DOI:10.1002/(SICI)1099-0518(199808)36:11<1685::AID-POLA1>3.0.CO;2-R |
| [59] |
C. Gao, D. Yan, Prog. Polym. Sci. 29 (2004) 183-275. DOI:10.1016/j.progpolymsci.2003.12.002 |
| [60] |
T. Sato, Y. Arima, M. Seno, T. Hirano, Macromolecules 38 (2005) 1627-1632. DOI:10.1021/ma040127z |
| [61] |
N. O'Brien, A. McKee, D.C. Sherrington, A.T. Slark, A. Titterton, Polymer 41 (2000) 6027-6031. DOI:10.1016/S0032-3861(00)00016-1 |
| [62] |
C.J. Hawker, J.M.J. Frechet, R.B. Grubbs, J. Dao, J. Am. Chem. Soc. 117 (1995) 10763-10764. DOI:10.1021/ja00148a027 |
| [63] |
C. Li, J. He, L. Li, J. Cao, Y. Yang, Macromolecules 32 (1999) 7012-7014. DOI:10.1021/ma990201r |
| [64] |
S.G. Gaynor, S. Edelman, K. Matyjaszewski, Macromolecules 29 (1996) 1079-1081. DOI:10.1021/ma9513877 |
| [65] |
C.Y. Hong, C.Y. Pan, Polymer 42 (2001) 9385-9391. DOI:10.1016/S0032-3861(01)00494-3 |
| [66] |
P.F.W. Simon, W. Radke, A.H.E. Müller, Macromol. Rapid Commun. 18 (1997) 865-873. DOI:10.1002/marc.1997.030180915 |
| [67] |
R. Francis, A. Ajayaghosh, Polymer 36 (1995) 1091-1096. DOI:10.1016/0032-3861(95)93611-O |
| [68] |
Z. Wang, J. He, Y. Tao, et al., Macromolecules 36 (2003) 7446-7452. DOI:10.1021/ma025673b |
| [69] |
C. Zhang, Y. Zhou, Q. Liu, et al., Macromolecules 44 (2011) 2034-2049. DOI:10.1021/ma1024736 |
| [70] |
X. Zhou, J. Zhu, M. Xing, et al., Eur. Polym. J. 47 (2011) 1912-1922. DOI:10.1016/j.eurpolymj.2011.07.002 |
| [71] |
J. Han, S. Li, A. Tang, C. Gao, Macromolecules 45 (2012) 4966-4977. DOI:10.1021/ma300718d |
| [72] |
A.H.E. Müller, D. Yan, M. Wulkow, Macromolecules 30 (1997) 7015-7023. DOI:10.1021/ma9619187 |
| [73] |
K. Min, H. Gao, J. Am. Chem. Soc. 134 (2012) 15680-15683. DOI:10.1021/ja307174h |
| [74] |
R.W. Graff, X. Wang, H. Gao, Macromolecules 48 (2015) 2118-2126. DOI:10.1021/acs.macromol.5b00278 |
| [75] |
R.W. Graff, Y. Shi, X. Wang, H. Gao, Macromol. Rapid Commun. 36 (2015) 2076-2082. DOI:10.1002/marc.201500388 |
| [76] |
X. Wang, R.W. Graff, Y. Shi, H. Gao, Polym. Chem. 6 (2015) 6739-6745. DOI:10.1039/C5PY01043H |
| [77] |
S. Misra, X. Wang, I. Srivastava, et al., Chem. Commun. (Camb.) 51 (2015) 16710-16713. DOI:10.1039/C5CC07709E |
| [78] |
L. Zhao, X. Wu, X. Wang, et al., ACS Macro Lett. 6 (2017) 700-704. DOI:10.1021/acsmacrolett.7b00242 |