 2019, Vol. 30
2019, Vol. 30 


Triplet-triplet annihilation upconversion (TTA-UC) has been attracting growing attention due to significant advantages in high upconversion quantum yield and ultra-low excitation intensities over other UC techniques, such as UC generated by rare earth metal materials and two-photon-absorption dyes, and has found substantial applications in photovoltaics [1, 2], bioimaging [3, 4], photocatalysis [5-8] and photoelectric devices [9]. A dye pair of socalled triplet sensitizer and acceptor/annihilator undergoes multistep energy transfer/fusion to complete the UC process [10]. In general, the photosensitizer is excited with low-energy photons, undergoes the intersystem crossing (ISC) to its triplet state, transfers the energy to the acceptor through triplet-triplet energy transfer (TTET), and the radiative transition from the singlet state acceptor arising from the TTA of two triplet state acceptors results in the UC fluorescence [11, 12]. Since both the TTET from a sensitizer to an acceptor and the TTA between two acceptors obey the Dexter energy transfer mechanism, free diffusion of UC components was considered to be critical for an efficient TTA-UC in homogeneous solution [13, 14]. Numerous efforts have been made to optimize the photophysical properties of the photosensitizers, e.g., prolonging the triplet lifetime of the sensitizer to assure a long diffusion distance in bulk solvent during the excited state, and increasing the molar extinction coefficient of the acceptor at the excitation wavelength to allow for the excitation power lower than the solar irradiance [15-19].
Up to now, most TTA-UC were realized in organic solvent, which, however, has obvious weakness due to their effumability, biological incompatibility and hazardness [20-23]. The research of TTA-UC in aqueous media has recently received increasing attention. However, the poor solubility of organic sensitizers and acceptors in water often results in aggregation of the UC components, and therefore causes inefficient energy transfer due to the reduced mobility of the triplet excitons. Furthermore, aggregation often significantly changes the photophysical properties of the UC components, such like aggregation-induced emission quenching. Hence, achieving efficient TTA-UC emission in aqueous solution is highly challenging [24-26]. We have recently reported a supramolecular host-guest complexation strategy to enhance the TTET and TTA-UC efficiency in organic solvent by drawing the sensitizer and annihilator in close proximity [27], and demonstrated that efficient TTA-UC in water can be achieved by the self-aggregation of water-soluble acceptors based on the γ-cyclodextrin-modified DPA [10]. Herein, we report enhancing TTA-UC by a synergistic effect of host-guest complexation of the sensitizer with the acceptor and the self-aggregation of the acceptor observed with a dye pair consisting of a permethylated-β- cyclodextrin (PMCD)-modified schiff-base Pt(II) complex and a 9, 10-diphenylanthracence (DPA) dimer.
PMCD derivative Pt-2 was fabricated by reacting 6A-aminoPMCD with a Schiff base platinum complex Pt-1 (Scheme 1). Cyclodextrins can complex a wide range of organic guest molecules with moderate binding affinity in aqueous solution and have found many unique applications as supramolecular host [28-33]. Since PMCD showed significant affinity with many aromatic compounds, such as porphyrin derivative [34-38], we expected that PMCD should play two important roles when grafting onto Pt-2: Firstly, it improves the solubility of the sensitizer in aqueous solution by virtue of the excellent water solubility of PMCD, and secondly, provides a hydrophobic cavity to bind the acceptor and hence to enhance the energy transfer efficiency by drawing the sensitizer and acceptor in close proximity through host-guest complexation. Acceptor A-1 was synthesized as an energy acceptor which could also be an ideal guest molecule for Pt-2. In order to improve TTA efficiencies, two DPA carboxylate were covalently linked through an alkyl chain to give A-2. A-2 was expected to have higher energy transfer efficiency due to intramolecular TTA instead of the intermolecular ones.

|
Download:
|
| Scheme 1. The chemical structures and synthetic routes of the complexes Pt-1, Pt-2, A-1 and A-2. Reagents and conditions: (ⅰ) 4-toluene sulfonyl chloride, pyridine, r.t., 2 h; (ⅱ) NaN3, H2O, 80 ℃, 4 h; (ⅲ) NaH, CH3I, DMF, 0 ℃, 50 min; (ⅳ) PPh3, acetone, 3 h; H2O, 1 h, r.t.; (ⅴ) EtOH, r.t., 2 h; (ⅵ) K2PtCl4, K2CO3, 80 ℃, 18 h; (ⅶ) HOBt, EDC, THF, r.t., 3 h; (ⅷ) K2CO3, CsF, Pd(PPh3)4, toluene/THF/H2O (4:6:1), 140 ℃, 10 h; (ⅸ) NaOH, 1, 4-dioxane/water (5:1), 90 ℃, 6 h; (ⅹ) K2CO3, CsF, Pd(PPh3)4, toluene/THF/H2O (4:6:1), 140 ℃, 10 h; (xi) 1, 4-diiodobutane, K2CO3, acetone, 80 ℃, 4 h; (xii) NaOH, 1, 4-dioxane/water (5:1), 90 ℃, 6 h. | |
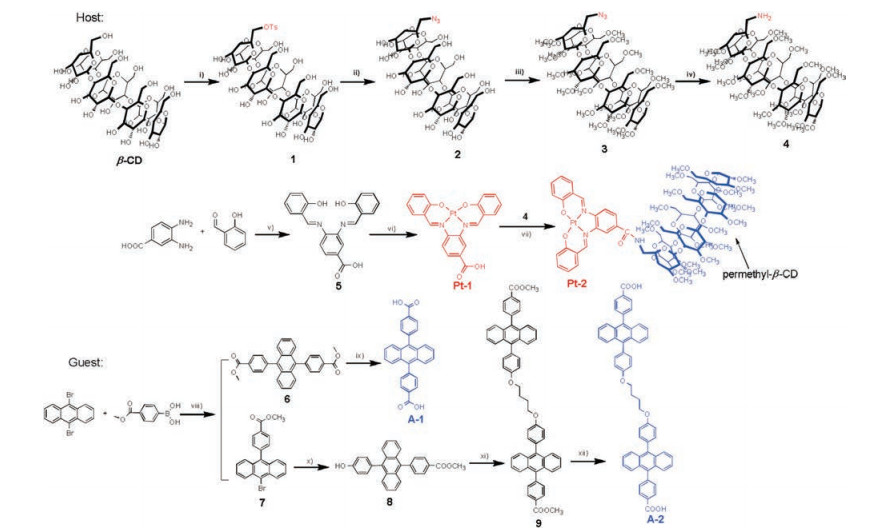
The absorption and emission spectra of the sensitizers and acceptors are shown in Fig. 1. Compound A-1 showed typical sharp peaks of DPA (Fig. S11 in Supporting information), while A-2 showed much blunter peaks and both absorption and emission wavelengths were red-shifted relative to compound A-1 (Fig. 1a), the molar extinction coefficient (ε) is smaller by two-fold than that of A-1 (Table 1). At the concentration of 10 μmol/L, the baseline of A-2 raised up at the longer wavelength, implying that A-2 is readily aggregate even if in DMSO, and the fluorescence quantum yield of A-2 is 54.1%, which is much lower than 81.7% for A-1. Both absorption and emission spectra of Pt-2 showed a little bit bathochromic-shift relative to that of Pt-1, probably due to the conjugation effect of amido group in Pt-2. The phosphorescence quantum yields and the triplet lifetime of Pt-2 are very similar to that of Pt-1, demonstrating that the introduction of PMCD did not significantly change the photophysical properties of the sensitizer.

|
Download:
|
| Fig. 1. Normalized UV–vis absorption and emission spectra of A-1, Pt-1 and Pt-2 in MeOH, and A-2 in DMSO, measured at 10 μmol/L at 25 ℃. | |
|
|
Table 1 Photophysical parameters of sensitizers and acceptors.a |

{kind=link}
{kind=link}
The host-guest interaction of PMCD and A-1 was investigated by UV–vis and 1H NMR titration method. Addition of PMCD to the solution of A-1 led to an increase of the absorption band at 300– 450 nm where PMCD showed no absorption (Fig. 2a), suggesting the binding of A-1 by PMCD. The 1H-1H COSY spectrum was measured to differentiate the aromatic protons of A-1 (Fig. S20 in Supporting information). 1H NMR titration experiment was further performed by fixing the concentration of A-1 and gradually increasing the concentration of PMCD. As illustrated in Fig. 2c, the chemical shifts of protons a, c and d in A-1 gradually shifted to the low field upon the addition of PMCD to the deuteroxide solution of A-1, while proton b shifted to the high field. At the same time, part of the protons in PMCD showed chemical shift to the low field. These phenomena undoubtedly demonstrating the host-guest complexation between PMCD and A-1. Moreover, the shift of A-1 proton signals stopped when the host-guest ratio reached 2:1, indicating a strong binding and it has a 2:1 host guest stoichiometry. By applying the nonlinear curve fitting method, the association constants assuming a stepwise formation of the 1:1 (K1) and 2:1 (K2) host-guest complex between PMCD and A-1 was determined to be K1 = 1.85×104 L/mol and K2 = 2.12×104 L/mol (Fig. 2b), which are much stronger than common complexation between native CD with organic guests [39-47]. The fluorescence titrations of A-2 with PMCD was measured to investigate the affinity of A-2 and PMCD (Fig. S22 in Supporting information), the binding constants was determined to be K1 = 2.70×105 L/mol and K2 = 2.30×105 L/mol, which is larger than that of A-1 with PMCD. The stronger affinity of A-2 with PMCD is probably due to the larger aromatic moiety of A-2 which results in the stronger hydrophobic interactions.

|
Download:
|
| Fig. 2. (a) UV–vis spectral changes of A-1 upon addition of various amounts of PMCD in water at room temperature, C[A-1] = 20 μmol/L. (b) The non-linear curve-fitting (UV– vis titrations) for the complexation of A-1 with PMCD, the association constants for the complex is K1 = 1.85(±0.47)×104 L/mol, K2 = 2.12 (0.47)×104 L/mol. (c) The chemical structures of A-1 and PMCD. (d) The 1H NMR spectra of the mixture of A-1 with different portions of PMCD in D2O. | |
{kind=link}
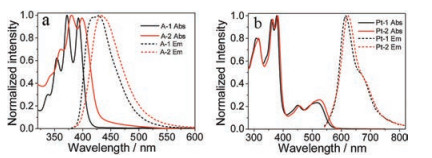
Since PMCD showed strong binding with A-1 and A-2, it is reasonable to expect that the energy transfer between Pt-2 and A-1/A-2 is efficient by virtue of the intimate contact of the Schiffbase Pt(II) complex and A-1/A-2 in the inclusion complex. We thus tried the upconversion experiments in water by using Pt-2 as the sensitizer and A-1/A-2 as the acceptor/annihilator. A 532 nm diode pump solid state (DPSS) laser was used as the exciting light source with Na2SO3 severing as a chemical deoxidant. Pt-2 itself emitted orange phosphorescence in the water. The addition of A-2 led to the quenching of the Pt-2 phosphorescence, accompanied by a blue emission at 400–500 nm, which is very similar with the emission of A-2 (Fig. S23 in Supporting information), while photoexcitation of A-2 with the 532 nm laser in the absent of Pt-2 did not result in any emission (Fig. 3a) in this wavelength range. The blue emission of A-2 increased with the concentration of A-2 and reached a maximum when the concentration was 40 μmol/L, with the maximal quantum yield being determined to be 0.1% in the 0.1 mol/L Na2SO3 aqueous solution.

|
Download:
|
| Fig. 3. (a) The emission spectra of Pt-2, A-2 and the mixture of Pt-2 and A-2 in the 0.1 mol/L Na2SO3 aqueous solution, CPt-2 = 2.5 μmol/L, CA-2 = 40 μmol/L, λex = 532 nm, at 318 mW/cm2 and 25 ℃. (b) Delayed fluorescence decay observed in the TTA upconversion. λex = 532 nm (nanosecond pulsed OPO laser synchronized with spectrofluorometer) and λem = 430 nm. (c) The upconversion spectra of A-1/A-2 with Pt-1/Pt-2 served as sensitizers in 0.1 mol/L Na2SO3 aqueous solution, CA-1 = CA-2 = 40 μmol/L, CPt-1 = CPt-2 = 2.5 μmol/L, λex = 532 nm, at 318 mW/cm2 and 25 ℃. (d) Double logarithmic plot of the UC emission intensity as a function of light power density. | |
{kind=link}
The relatively low UC quantum yield could be partly attributed to the decreased fluorescence quantum yield of A-2 (Table 1). In order to elucidate the nature of the blue emission observed in the aqueous solution containing Pt-2 and A-2, the emission decay of the blue emission excited with a nanosecond-pulsed OPO laser was measured. The lifetime of the emission peaked at 430 nm was determined to be 16.6 ms (Fig. 3b), which is overwhelmingly longer than the lifetime of the prompt fluorescence of A-2 (3.3 ns, Table 1). This indicated that the blue emission arised from the TTA upconversion. The emission intensity of A-2 as a function of the excitation power density was further investigated. The UC emission increased with the excitation power (Fig. S18 in Supporting information), and the double logarithmic plots of UC intensity versus the light power density followed firstly a quadratic process and then switched to a linear process when the power density increase to 232 mW/cm2 (Fig. 3d), indicating that TTA become a dominant decay path of the triplet states of A-2 after this threshold value. The quadratic relationship of the intensity versus excitation power density further verified the TTA based upconversion mechanism.
Interestingly, much weaker UC emission was observed when Pt-1 and A-2 were used as the donor/acceptor pair comparing with Pt-2/A-2 system (Fig. 3c), indicating the important roles played by the host-guest interaction between PMCD and A-2. However, very weak upconversion emission was observed when Pt-2 and A-1 was used as the dye pair (Fig. 3c). This is slightly unexpected, as strong host-guest interaction between Pt-2 and A-1 should cause efficient triplet-triplet energy transfer between Pt-2 and A-1. Moreover, A-1 showed much higher fluorescence quantum yield than A-2, which should also be beneficial for efficient TTA-UC. The blue upconverted emission of Pt-2/A-2 system is visible with a short wave pass filter (Fig. S19 in Supporting information).
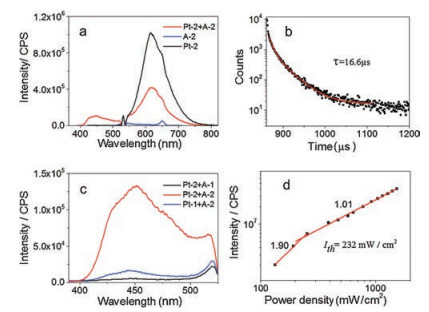
To further understand the origin of the stronger UC emission with A-2, the aggregation behavior of acceptors A-1 and A-2 was investigated. Absorption spectra of both A-1 and A-2 were measured at different concentrations. As shown in Fig. 4, the baseline of the absorption spectra of the acceptor A-2 raised up obviously when increasing the concentration from 2 μmol/L to 40 μmol/L in aqueous solution (Fig. 4a), while the absorption spectra of acceptor A-1 was hardly changed at the same concentration range (Fig. 4b), implying an aggregation of A-2 in the aqueous solution. This seems reasonable because the larger aromatic moiety of A-2 should bring stronger hydrophobic and π–π stacking interactions. Indeed, A-1 showed much better water solubility under the alkaline conditions. Dynamic light scattering (DLS) studies revealed that A-2 at the concentration of 2.5 μmol/L afforded nanostructures of a mean size of 526 nm, while micronscale aggregates of 3.6 mm was formed by increasing the concentration to 40 μmol/L (Fig. 4c). The aggregations of A-2 was further confirmed by means of scanning electron microscope (SEM), and long microstrips with widths of several to tens micrometers was observed from the aggregation generated from 40 μmol/L aqueous solution (Fig. 4d).

|
Download:
|
| Fig. 4. The normalized UV–vis spectra of (a) A-2 and (b) A-1, respectively, at different concentration in aqueous buffer solution at room temperature. (c) The DLS spectra of compound A-2 at 2.5 μmol/L and 40 μmol/L, respectively, in aqueous buffer solution measured at 25 ℃. (d) The SEM images of A-2 generated at 40 μmol/L in aqueous solution. | |
{kind=link}
Considering the fact that A-2 showed significant selfaggregation in aqueous solution and PMCD showed high affinity with DPA carboxylate, we attribute the much stronger upconverted emission of Pt-2/A-2 pair to the synergistic effect of host-guest complexation and self-assembling of the acceptor. As represented in Fig. 5, complexation of Pt-2 with A-2 led to efficient TTET from the Schiff base Pt complex to DPA unit. On the other hand, the aggregation of A-2 should facilitate the triplet energy transfer among acceptors through triplet exciton migration [48, 49], so as to improve both the TTET and TTA processes. The much lower TTA-UC observed with Pt-1/A-2 pair could thus be ascribed to the lack of host-guest complexation and relatively low solubility of Pt-1, which readily aggregates and has to diffuse and collide the assemblies of A-2 to accomplish the energy transfer. While in the case of Pt-2/A-1 pair, most of the acceptors at the triplet excited state should be complexed by Pt-2 due to the much more efficient intra-complex energy transfer and the bulky complex of Pt-2 and A-1 should have much lower movability than free A-1.

|
Download:
|
| Fig. 5. The schematic UC emission mechanism with Pt-1/Pt-2 as the sensitizers and A-1/ A-2 as the acceptors. | |
{kind=link}
In conclusion, a dye pair consisting of a permethyl-β-CD appending Schiff-base Pt complex (Pt-2) and a DPA dimer (A-2) was devised for improving TTA upconversion in aqueous solution. Permethyl-β-CD showed high affinity with the DPA carboxylate with a binding constant of 1.85×104 L/mol, which guaranteed efficient TTET from the Schiff-base Pt complex unit in Pt-2 to the included DPA units of A-2. Aggregation of the dimer A-2 in aqueous solution allowed for efficient triplet energy migration among the acceptor, and thus, improved TTA upconverison (ΦUC = 0.1%) was observed. Such a TTA-UC quantum yield is significantly stronger than other dye pairs (Pt-2/A-1 and Pt-1/A-2), in which the hostguest complexation between the sensitizer and the acceptor and self-assembling of the acceptor did not cooccur. This work provides a new strategy for improving the TTA-UC efficiency in aqueous solution and has the potential for the applications in molecular sensing based on supramolecular TTA-UC.
AcknowledgmentsWe acknowledge the financial support of the National Natural Science Foundation of China (Nos. 21971169, 21871194, 21572142, 21372165 and 21321061), National Key Research and Development Program of China (No. 2017YFA0505903), and Science & Technology Department of Sichuan Province (Nos. 2019YJ0090, 2019YJ0160, 2017SZ0021). Comprehensive Training Platform of Specialized Laboratory, College of Chemistry and Prof. Peng Wu of Analytical & Testing Center, Sichuan University for characterization and lifetime measurement are greatly appreciated.
Appendix A. Supplementary dataSupplementary material related tothis article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.09.009.
| [1] |
Y.Y. Cheng, B. Fückel, R.W. MacQueen, et al., Energy Environ. Sci. 5 (2012) 6953-6959. DOI:10.1039/c2ee21136j |
| [2] |
A. Monguzzi, D. Braga, M. Gandini, et al., Nano Lett. 14 (2014) 6644-6650. DOI:10.1021/nl503322a |
| [3] |
J. Wang, Y. Lu, N. McGoldrick, et al., J. Mater. Chem. C 4 (2016) 6131-6139. |
| [4] |
O.S. Kwon, H.S. Song, J.O. Conde, et al., ACS Nano 10 (2016) 1512-1521. DOI:10.1021/acsnano.5b07075 |
| [5] |
J.I. Goldsmith, W.R. Hudson, M.S. Lowry, et al., J. Am. Chem. Soc. 127 (2005) 7502-7510. DOI:10.1021/ja0427101 |
| [6] |
R.R. Islangulov, F.N. Castellano, Angew. Chem. Int. Ed. 45 (2006) 5957-5959. DOI:10.1002/anie.200601615 |
| [7] |
O.S. Wenger, Chem.-Eur. J. 17 (2011) 11692-11702. DOI:10.1002/chem.201102011 |
| [8] |
M. Rao, K. Kanagaraj, C. Fan, et al., Org. Lett. 20 (2018) 1680-1683. DOI:10.1021/acs.orglett.8b00520 |
| [9] |
C.H. Chen, N.T. Tierce, M.K. Leung, et al., Adv. Mater. 30 (2018) 1804850.
|
| [10] |
W. Xu, W. Liang, W. Wu, et al., Chem.-Eur. J. 24 (2018) 16677-16685. DOI:10.1002/chem.201804001 |
| [11] |
T.N. Singh-Rachford, F.N. Castellano, Coord. Chem. Rev. 254 (2010) 2560-2573. DOI:10.1016/j.ccr.2010.01.003 |
| [12] |
C. Ye, L. Zhou, X. Wang, et al., PCCP 18 (2016) 10818-10835. DOI:10.1039/C5CP07296D |
| [13] |
T. Ogawa, M. Hosoyamada, B. Yurash, et al., J. Am. Chem. Soc. 140 (2018) 8788-8796. DOI:10.1021/jacs.8b04542 |
| [14] |
P. Bharmoria, S. Hisamitsu, H. Nagatomi, et al., J. Am. Chem. Soc. 140 (2018) 10848-10855. DOI:10.1021/jacs.8b05821 |
| [15] |
Y. Che, W. Yang, G. Tang, et al., J. Mater. Chem. C 6 (2018) 5785-5793. DOI:10.1039/C8TC00513C |
| [16] |
Y. Bai, J.H. Olivier, H. Yoo, et al., J. Am. Chem. Soc. 139 (2017) 16946-16958. DOI:10.1021/jacs.7b09982 |
| [17] |
Z. Wang, J. Zhao, A. Barbon, et al., J. Am. Chem. Soc. 139 (2017) 7831-7842. DOI:10.1021/jacs.7b02063 |
| [18] |
H. Jia, B. Küçüköz, Y. Xing, et al., J. Mater. Chem. C 2 (2014) 9720-9736. DOI:10.1039/C4TC01675K |
| [19] |
W. Wu, J. Zhao, J. Sun, et al., J. Mater. Chem. C 1 (2013) 705-716. DOI:10.1039/C2TC00214K |
| [20] |
J. Park, M. Xu, F. Li, et al., J. Am. Chem. Soc. 140 (2018) 5493-5499. DOI:10.1021/jacs.8b01613 |
| [21] |
H. Kouno, T. Ogawa, S. Amemori, et al., Chem. Sci. 7 (2016) 5224-5229. DOI:10.1039/C6SC01047D |
| [22] |
K. Tanaka, K. Inafuku, Y. Chujo, Chem. Commun. 46 (2010) 4378-4380. DOI:10.1039/c0cc00266f |
| [23] |
Q. Liu, M. Xu, T. Yang, et al., ACS Appl. Mater. Interface 10 (2018) 9883-9888. |
| [24] |
C. Kerzig, O.S. Wenger, Chem. Sci. 9 (2018) 6670-6678. DOI:10.1039/C8SC01829D |
| [25] |
H. Kouno, Y. Sasaki, N. Yanai, et al., Chem.-Eur. J. 25 (2019) 6124-6130. DOI:10.1002/chem.201806076 |
| [26] |
H.Q. Peng, L.Y. Niu, Y.Z. Chen, et al., Chem. Rev. 115 (2015) 7502-7542. DOI:10.1021/cr5007057 |
| [27] |
C. Fan, W. Wu, J.J. Chruma, et al., J. Am. Chem. Soc. 138 (2016) 15405-15412. DOI:10.1021/jacs.6b07946 |
| [28] |
M.V. Rekharsky, Y. Inoue, Chem. Rev. 98 (1998) 1875-1918. DOI:10.1021/cr970015o |
| [29] |
X. Yu, W. Liang, Q. Huang, et al., Chem. Commun. 55 (2019) 3156-3159. DOI:10.1039/C9CC00097F |
| [30] |
J. Ji, W. Wu, W. Liang, et al., J. Am. Chem. Soc. 141 (2019) 9225-9238. DOI:10.1021/jacs.9b01993 |
| [31] |
X. Wei, W. Wu, R. Matsushita, et al., J. Am. Chem. Soc. 140 (2018) 3959-3974. DOI:10.1021/jacs.7b12085 |
| [32] |
J. Yi, W. Liang, X. Wei, et al., Chin. Chem. Lett. 29 (2018) 87-90. DOI:10.1016/j.cclet.2017.05.004 |
| [33] |
Z. Yan, Q. Huang, W. Liang, et al., Org. Lett. 19 (2017) 898-901. DOI:10.1021/acs.orglett.7b00057 |
| [34] |
Z.Y. Gu, D.S. Guo, M. Sun, et al., J. Org. Chem. 75 (2010) 3600-3607. DOI:10.1021/jo100351f |
| [35] |
Z.Q. Li, Y.M. Zhang, D.S. Guo, et al., Chem.-Eur. J. 19 (2013) 96-100. DOI:10.1002/chem.201203575 |
| [36] |
Y. Yang, Y.M. Zhang, Y. Zhang, et al., Chem. Asian J. 10 (2015) 84-90. DOI:10.1002/asia.201402802 |
| [37] |
G. Liu, X. Xu, Y. Chen, et al., Chem. Commun. 52 (2016) 7966-7969. DOI:10.1039/C6CC02996E |
| [38] |
X. Chen, Y.N. Wang, R.X. Rong, et al., Dyes Pigm. 160 (2019) 779-786. DOI:10.1016/j.dyepig.2018.09.011 |
| [39] |
R. Liu, Y. Zhang, W. Wu, et al., Chin. Chem. Lett. 30 (2019) 577-581. DOI:10.1016/j.cclet.2018.12.002 |
| [40] |
Q. Huang, L. Jiang, W. Liang, et al., J. Org. Chem. 81 (2016) 3430-3434. DOI:10.1021/acs.joc.6b00130 |
| [41] |
C. Yang, Q. Wang, M. Yamauchi, et al., Photochem. Photobiol. Sci. 13 (2014) 190-198. DOI:10.1039/C3PP50255D |
| [42] |
M. Nishijima, M. Goto, M. Fujikawa, et al., Chem. Commun. 50 (2014) 14082-14085. DOI:10.1039/C4CC04818K |
| [43] |
J. Yao, Z. Yan, J. Ji, et al., J. Am. Chem. Soc. 136 (2014) 6916-6919. DOI:10.1021/ja5032908 |
| [44] |
W. Liang, C. Yang, D. Zhou, et al., Chem. Commun. 49 (2013) 3510-3512. DOI:10.1039/c3cc40542g |
| [45] |
L. Dai, W. Wu, W. Liang, et al., Chem. Commun. 54 (2018) 2643-2646. DOI:10.1039/C8CC00840J |
| [46] |
X. Wei, X. Yu, Y. Zhang, et al., J. Photochem. Photobio. A:Chem. 371 (2019) 374-381. DOI:10.1016/j.jphotochem.2018.11.038 |
| [47] |
M. Rao, W. Wu, C. Yang, Molecules 24 (2019) 1502.
|
| [48] |
N. Kimizuka, N. Yanai, M.A. Morikawa, Langmuir 32 (2016) 12304-12322. DOI:10.1021/acs.langmuir.6b03363 |
| [49] |
N. Yanai, N. Kimizuka, Chem. Commun. 52 (2016) 5354-5370. DOI:10.1039/C6CC00089D |