 2019, Vol. 30
2019, Vol. 30 


b Department of Materials Chemistry, Huzhou University, Huzhou 313000, China;
c Shanxi Institute of Flexible Electronics(SIFE), Northwestern Polytechnical University(NPU), Xi'an 710072, China
Ultralong organic phosphorescence (UOP) in metal-free luminescent materials has invoked popular research interests in various fields including sensing, bioimaging, encryption and so forth [1-9]. However, with the absence of heavy metal atoms, exploring efficient UOP materials is extremely difficult due to the inefficient intersystem crossing (ISC) caused by weak spin-orbit coupling (SOC), and the ultrafast non-radiative decay pathways of the triplet excitons through molecular motions, humidity or oxygen quenching from the surroundings and so on [10-13]. To overcome these obstacles, tremendous efforts have been made to enhance the ISC rate through the introduction of heavy atoms or aromatic carbonyl groups [14-18], or to suppress nonradiative relaxation via H-aggregation [19-21], host-guest doping methods [22-26], crystallization inducement [27-36], organic framework construction and polymerization [37-45]. Despite various strategies have been put forward to facilitate UOP, it is still a challenge to obtain highly efficient UOP in metal-free organic luminogens under ambient conditions.
The well-known photophysical process of phosphorescence quantum yield (Φp) and lifetime (τp) can be expressed by the equations shown in Fig. 1, where kr and knr are the rate constants for phosphorescence radiative and nonradiative deactivations from the T1 to S0 state, respectively [46-49]. Based on the equations, it is rational to expect that modulating kr and knr values can realize the UOP properties manipulation. However, there is a general problem that enhancing ISC via promoting spin-orbit coupling will accelerate the kr of T1 at the same time lead to a short lifetime. In this case, how to reasonably tune the kr and knr to achieve highly efficient UOP in metal-free organic molecules is meaningful but difficult. To this end, we proposed a facile way of subtle structure tailoring in metal-free triazine luminogens.

|
Download:
|
| Fig. 1. Schematic illustration for achieving highly efficient UOP materials and chemical structures of o-BrAT, m-BrAT and p-BrAT isomers. | |
{kind=link}
Herein, we designed and synthesized three isomers based on a series of asymmetric triazines with various Br-substituted positions, namely o-BrAT, m-BrAT and p-BrAT (Fig. 1). It was found that all the isomers displayed ultralong phosphorescence characteristics under ambient conditions, wherein o-BrAT in the solid state exhibited the longest phosphorescent lifetime of 792 ms, three times higher than that of m-BrAT owing to effective intermolecular interactions. Notably, phosphorescence efficiency of p-BrAT in solid can reach up to 9.7% with a long lifetime of 386 ms, achieving an impressive highly efficient UOP property. Combining X-ray single crystal analysis with theoretical calculations, it proved that the one order of magnitude rising of kr value in p-BrAT crystal might play a significant role in improving phosphorescence efficiency, which was consistent with the calculated oscillator strength (f) values.
Three isomers were readily prepared through two-step reactions (Scheme S1 in Supporting information) with yields of 29.4%, 35.0% and 28.5% for o-BrAT, m-BrAT and p-BrAT, respectively. The chemical structures and purities of the target compounds were thoroughly characterized by NMR spectroscopy (Figs. S1-S3 in Supporting information), matrix-assisted laser desorption/ionization time of flight mass spectrometry (MALDI-TOF MS), elemental analysis and X-ray single crystal analysis. The single-crystal data showed that the variation of bromine atom substituted positions had little influence on the molecular configuration but caused different crystal stacking models, resulting in different intermolecular interactions for the three isomers in crystal.
The photophysical properties of these compounds were systematically investigated in solution by absorption and steady-state photoluminescence (PL) spectra firstly. In dilute chloroform solution (1 ×10-5 mol/L), the absorption spectra of three isomers were completely consistent with the peaks at 308 and 321 nm (Fig. S4a in Supporting information). Accordingly, their steady-state PL spectra in dilute solution excited by 320 nm were identical with a broad emission band at around 450 nm, which was attributed to charge transfer (CT) state emission owing to the existence of donor-acceptor (D-A) structures in these isomers [50, 51]. The lifetimes of emission bands at around 450 nm were 1.42, 1.43 and 1.45 ns for o-BrAT, m-BrAT and p-BrAT in solution, respectively (Fig. S5 in Supporting information). Besides, the quantum yields for o-BrAT, m-BrAT and p-BrAT in solution were 2.6%, 3.5% and 3.0%, respectively. Interestingly, at 77 K, the PL and phosphorescence spectra of the three compounds almost reached superposition (Fig. S4b in Supporting information), which indicated the nearly 100% ISC efficiency of singlet excitons. Meanwhile, the profiles of their phosphorescence spectra were identical, indicating that the phosphorescence probably was resulted from the same carbazole chromophore. These results demonstrated that subtle structure tailoring of heavy atom substituted positions in these isomers had little impact on their photophysical properties in single molecule state.
Impressively, the three compounds in solid state all displayed yellow bright afterglow emission, which can last for 1–5 s after the UV-light was turned off (SV1-SV3). Therefore, a set of experiments on the photophysical properties of the three compounds in crystal state were carried out. As shown in Fig. 2a, their PL spectra showed obvious fluorescence bands at 390–430 nm excited by 350 nm under ambient condition. Moreover, m-BrAT showed a weak extra emission band at 520–600 nm, while p-BrAT exhibited intense extra emission bands at 533 and 577 nm and a shoulder at 630 nm upon 350 nm excitation. Owing to the different emission contribution in long wavelength intervals, their emission showed blue, blue-white and white colors for o-BrAT, m-BrAT and p-BrAT, respectively under 365 nm excitation, as shown in the luminescent images. Phosphorescence spectra of these isomers all showed similar profiles with a major peak at around 530 nm and two shoulders at around 570 and 630 nm. As depicted in the timeresolved emission decay curves (Figs. 2b and c and Fig. S6 in Supporting information), the phosphorescence lifetimes at main emission bands for o-BrAT, m-BrAT and p- BrAT were 792, 187 and 386 ms, respectively, indicating their UOP properties. The phosphorescence nature of emission was further confirmed by the oxygen-dependent phosphorescence experiments (Fig. S7 in Supporting information) [52, 53]. In the further investigation, their PL quantum yields (QEs) were measured using a fluorescence spectrophotometer equipped with an integrating sphere under ambient conditions. As shown in Fig. 2b and Table S1 (Supporting information), o-BrAT in solid had relatively low phosphorescence but high fluorescence QE, while m-BrAT had the lowest phosphorescence as well as fluorescence QE. Impressively, p-BrAT in solid exhibited the highest phosphorescence QE of up to 9.7%. To the best of our knowledge, it is one of the most highly efficient UOP materials reported [15, 54-58]. It is noteworthy that we achieved the simultaneous enhancement of phosphorescence lifetime and efficiency from m- to p-BrAT at room temperature, which was not commonly-seen in the reported cases. In addition, the phosphorescence spectra of p-BrAT in solid state remained at an unchanged emission position with excitation wavelengths varied from 250 nm to 390 nm, during which the phosphorescence emission intensity reached the strongest under 368 nm excitation (Fig. 2d). Therefore, based on such asymmetrical isomers via bromine placement variation, we obtained highly efficient UOP in metal-free luminogens.

|
Download:
|
| Fig. 2. Photophysical properties of o-BrAT, m-BrAT and p-BrAT molecules in solid state under ambient conditions. (a) Normalized steady-state photoluminescence (black dotted line) and phosphorescence (red solid line) spectra of three isomers excited by 350 nm. The insets are the photographs of the compounds in crystal state under the 365 nm UV light on (left) and off (right). (b) Variation tendency of phosphorescence quantum yield and lifetime for the three isomers. (c) Lifetime decay profiles of the emission bands at 533 nm and 576 nm for o-BrAT excited by 365 nm. (d) Excitation-phosphorescence mapping of p-BrAT powder. | |
{kind=link}
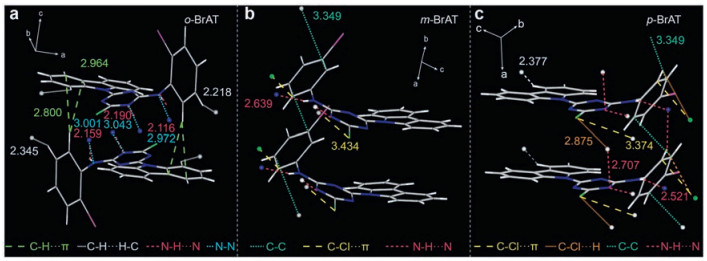
To get deeper insight into the mechanism for UOP generation of these isomers, both single crystal analysis and theoretical calculations were conducted. In crystal state, there exists strong π…π stacking between two carbazole units with the distances of 3.334, 3.476 and 3.529 Å for o-BrAT, m-BrAT and p-BrAT, respectively, which can facilitate electron communication between dimers. Notably, abundant and short intermolecular interactions including C-H…π (2.763, 2.830, 2.785, 2.765 Å), N-H…N (2.159, 2.190 Å), N…N (3.001, 3.043 Å), C-H…H-C (2.293 Å), and C…C (3.360 Å) are present in o-BrAT crystal, which formed a firm 3D network (Fig. 3a and Fig. S8 in Supporting information) to restrain the non-radiative transition effectively. Moreover, the multiple interactions are evenly distributed on every group of o-BrAT containing the carbazole chromophore, which is powerful to construct a rigid environment to decrease nonradiative transitions and endow o-BrAT with the longest phosphorescence lifetime. Similarly, p-BrAT in crystal can also form multiple interactions (Fig. 3c), but the molecular motions of carbazole chromophore cannot be restricted as well as o-BrAT and the crystal stacking is not so dense (Fig. S9 in Supporting information). Therefore, p-BrAT has a shorter UOP lifetime than o-BrAT. By contrast, there are fewer short contacts around m-BrAT molecule as well as a looser crystal stacking in the single crystal (Fig. 3b and Fig. S10 in Supporting Information). Moreover, there exists no short interactions on carbazole chromophore, which will increase the nonradiative transitions, resulting in the shortest UOP lifetime of m-BrAT. Moreover, the dihedral angles between the transition dipoles and interconnected axis measured for the three isomers are 64.6°, 55.4° and 74.4°, respectively, which all exceeded the critical angle 54.7° of H-aggregation (Fig. S11 in Supporting information). Thus, we reasoned that the existence of H-aggregation could stabilize the triplet excitons for ultralong phosphorescence in crystal under ambient conditions [19].

|
Download:
|
| Fig. 3. Molecular configuration and intramolecular interactions of (a) o-BrAT, (b) m-BrAT and (c) p-BrAT dimers in the single crystal state, respectively. | |
{kind=link}
To further understand the high phosphorescence quantum efficiency of p-BrAT, we performed first-principle time-dependent density functional theory (TD-DFT) of these isomers based on their single crystal dada. As illustrated in Table S2 (Supporting information) and Fig. 4, position isomerism has slight impact on the spin-orbit coupling (SOC) and excitation energy of T1 state (ΔE), however, it dramatically influenced the oscillator strengthen (f) with one order of magnitude raises in p-BrAT. According to the photophysical data of these isomers, we calculated their radiative and nonradiative decay rates (Table S3 in Supporting information). We then inferred that the hugely increased kr ∝ ΔE2f value (Fig. 4b) is responsible for the resulted high phosphorescence efficiency up to 9.7%, with assist of the slight changed knr ∝ SOC2exp(-ΔE2), which is also consistent with the variation tendency of these isomers in experiments (Figs. 4c and d).

|
Download:
|
| Fig. 4. (a) Excitation energy ΔE, oscillator strength (f) and spin-orbit coupling (SOC) of T1→S0 for the three isomers. (b) A Jablonski diagram representing the photophysical processes leading to highly efficient UOP for p-BrAT. The dependency of (c) phosphorescence efficiency and (d) lifetime with kr and knr, respectively. | |
{kind=link}
In summary, we designed three isomers with highly efficient UOP based on asymmetric triazine derivatives with slight tuning of bromine substituted positions. X-ray single crystal analysis indicates that different bromine positions has a dramatic influence on molecular stacking, thus effectively manipulating the phosphorescence lifetime and efficiency. Impressively, p-BrAT in solid state displayed a high phosphorescence efficiency up to 9.7% with a long lifetime of 386 ms, which was one of the highest efficient UOP materials reported so far in metal-free compounds. Taken theoretical calculations and experimental data, we concluded that the highly efficient UOP was ascribed to effective radiative transitions and stabilization of triplet excitons by H-aggregation. These results will provide an effective approach for achieving highly efficient UOP in metal-free materials, and will be promising in the potential applications in organic electronics and bioelectronics.
AcknowledgmentsThis work is supported by the National Natural Science Foundation of China (Nos. 21875104 and 51673095), National Basic Research Program of China (973 Program, No. 2015CB932200), Natural Science Fund for Distinguished Young Scholars (No. BK20180037), and the Natural Science Fund for Colleges and Universities (No. 17KJB430020), "High-Level Talents in Six Industries" (No. XCL-025) of Jiangsu Province and Nanjing Tech Start-up Grant (Nos. 3983500158 and 3983500169). We are grateful to the High Performance Computing Centre of Nanjing Tech University for supporting the computational resources.
Appendix A. Supplementary dataSupplementary material related to this article canbefound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2018.12.023.
| [1] |
R. Kabe, N. Notsuka, K. Yoshida, C. Adachi, Adv. Mater. 28 (2016) 655-660. DOI:10.1002/adma.201504321 |
| [2] |
X. Zhen, Y. Tao, Z. An, et al., Adv. Mater. 29 (2017) 1606665. DOI:10.1002/adma.201606665 |
| [3] |
S.M.A. Fateminia, Z. Mao, S. Xu, et al., Angew. Chem. Int. Ed. 56 (2017) 12160-12164. DOI:10.1002/anie.201705945 |
| [4] |
L. Gu, H. Shi, M. Gu, et al., Angew. Chem. Int. Ed. 57 (2018) 8425-8431. DOI:10.1002/anie.201712381 |
| [5] |
Z. Yu, Y. Wu, L. Xiao, et al., J. Am. Chem. Soc. 139 (2017) 6376-6381. DOI:10.1021/jacs.7b01574 |
| [6] |
K. Li, L. Zhao, Y. Gong, et al., Sci. China Chem. 60 (2017) 806-812. DOI:10.1007/s11426-016-0460-8 |
| [7] |
Y. Yang, K.Z. Wang, D. Yan, ACS Appl. Mater. Interfaces 9 (2017) 17399-17407. DOI:10.1021/acsami.7b00594 |
| [8] |
Q. Wu, H. Ma, K. Ling, et al., ACS Appl. Mater. Interfaces 10 (2018) 33730-33736. DOI:10.1021/acsami.8b13713 |
| [9] |
L. Huang, B. Chen, X. Zhang, et al., Angew. Chem. Int. Ed. 57 (2018) 16046-16050. DOI:10.1002/anie.201808861 |
| [10] |
H. Xu, R. Chen, Q. Sun, et al., Chem. Soc. Rev. 43 (2014) 3259-3302. DOI:10.1039/C3CS60449G |
| [11] |
Z. Pan, Y. Lu, F. Liu, Chem. Soc. Rev. 46 (2017) 275-299. DOI:10.1039/C6CS00551A |
| [12] |
E.M. Schulman, R.T. Parker, J. Phys. Chem. 81 (1977) 1932-1939. DOI:10.1021/j100535a010 |
| [13] |
M.A. El-Sayed, Acc. Chem. Res. 1 (1968) 8-16. DOI:10.1021/ar50001a002 |
| [14] |
Y. Xie, Y. Ge, Q. Peng, et al., Adv. Mater. 29 (2017) 1606829. DOI:10.1002/adma.201606829 |
| [15] |
Y. Xiong, Z. Zhao, W. Zhao, et al., Angew. Chem. Int. Ed. 57 (2018) 7997-8001. DOI:10.1002/anie.201800834 |
| [16] |
O. Bolton, K. Lee, H.J. Kim, et al., Nat. Chem. 3 (2011) 205. DOI:10.1038/nchem.984 |
| [17] |
H. Ma, W. Shi, J. Ren, et al., J. Phys. Chem. Lett. 7 (2016) 2893-2898. DOI:10.1021/acs.jpclett.6b01156 |
| [18] |
S. Kuila, K.V. Rao, S. Garain, et al., Angew. Chem. Int. Ed. 57 (2018) 17115-17119. DOI:10.1002/anie.201810823 |
| [19] |
Z. An, C. Zheng, Y. Tao, et al., Nat. Mater. 14 (2015) 685-690. DOI:10.1038/nmat4259 |
| [20] |
E. Lucenti, A. Forni, C. Botta, et al., J. Phys. Chem. Lett. 8 (2017) 1894-1898. DOI:10.1021/acs.jpclett.7b00503 |
| [21] |
S. Cai, H. Shi, J. Li, et al., Adv. Mater. 29 (2017) 1701244. DOI:10.1002/adma.201701244 |
| [22] |
S. Hirata, K. Totani, J. Zhang, et al., Adv. Funct. Mater. 23 (2013) 3386-3397. DOI:10.1002/adfm.201203706 |
| [23] |
C. Xu, L. Xu, X. Ma, Chin. Chem. Lett. 29 (2018) 970-972. DOI:10.1016/j.cclet.2017.11.045 |
| [24] |
T. Ogoshi, H. Tsuchida, T. Kakuta, et al., Adv. Funct. Mater. 28 (2018) 1707369. DOI:10.1002/adfm.201707369 |
| [25] |
S. Hirata, M. Vacha, J. Phys. Chem. Lett. 7 (2016) 1539-1545. DOI:10.1021/acs.jpclett.6b00554 |
| [26] |
D. Li, F. Lu, J. Wang, et al., J. Am. Chem. Soc. 140 (2018) 1916-1923. DOI:10.1021/jacs.7b12800 |
| [27] |
J. Yang, X. Zhen, B. Wang, et al., Nat. Commun. 9 (2018) 840-850. DOI:10.1038/s41467-018-03236-6 |
| [28] |
Z. Cheng, H. Shi, H. Ma, et al., Angew. Chem. Int. Ed. 57 (2018) 678-682. DOI:10.1002/anie.201710017 |
| [29] |
C. Sun, X. Ran, X. Wang, et al., J. Phys. Chem. Lett. 9 (2018) 335-339. DOI:10.1021/acs.jpclett.7b02953 |
| [30] |
C.R. Wang, Y.Y. Gong, W.Z. Yuan, Y.M. Zhang, Chin. Chem. Lett. 27 (2016) 1184-1192. DOI:10.1016/j.cclet.2016.05.026 |
| [31] |
W. Luo, Y. Zhang, Y. Gong, et al., Chin. Chem. Lett. 29 (2018) 1533-1536. DOI:10.1016/j.cclet.2018.08.001 |
| [32] |
W. Zhao, Z. He, J.W.Y. Lam, et al., Chem 1 (2016) 592-602. DOI:10.1016/j.chempr.2016.08.010 |
| [33] |
J. Wei, B. Liang, R. Duan, Angew. Chem. Int. Ed. 55 (2016) 15589-15593. DOI:10.1002/anie.201607653 |
| [34] |
G. Chen, H. Feng, F. Feng, et al., J. Phys. Chem. Lett. 9 (2018) 6305-6311. DOI:10.1021/acs.jpclett.8b02742 |
| [35] |
B. Zhou, D. Yan, Adv. Funct. Mater. 29 (2019) 1807599. DOI:10.1002/adfm.201807599 |
| [36] |
H. Liu, Z. Bian, Q. Cheng, et al., Chem. Sci. 10 (2019) 227-232. DOI:10.1039/C8SC03135E |
| [37] |
X. Yang, D. Yan, Chem. Sci. 7 (2016) 4519-4526. DOI:10.1039/C6SC00563B |
| [38] |
H. Mieno, R. Kabe, N. Notsuka, Adv. Optical Mater. 4 (2016) 1015-1021. DOI:10.1002/adom.201600103 |
| [39] |
X. Yang, D. Yan, Adv. Optical Mater. 4 (2016) 897-905. DOI:10.1002/adom.201500666 |
| [40] |
S. Cai, H. Shi, Z. Zhang, et al., Angew. Chem. Int. Ed. 57 (2018) 4005-4009. DOI:10.1002/anie.201800697 |
| [41] |
X. Chen, C. Xu, T. Wang, et al., Angew. Chem. Int. Ed. 55 (2016) 9872-9876. DOI:10.1002/anie.201601252 |
| [42] |
W. Shi, J. Yao, L. Bai, et al., Adv. Funct. Mater. 28 (2018) 1804961. DOI:10.1002/adfm.201804961 |
| [43] |
Y. Su, S.Z.F. Phua, et al., Sci. Adv. 4 (2018) EAAS9732. DOI:10.1126/sciadv.aas9732 |
| [44] |
T. Ogoshi, H. Tsuchida, T. Kakuta, et al., Adv. Funct. Mater. 28 (2018) 1707369. DOI:10.1002/adfm.201707369 |
| [45] |
N. Gan, H. Shi, Z. An, et al., Adv. Funct. Mater. 28 (2018) 1802657. DOI:10.1002/adfm.201802657 |
| [46] |
Y. Gong, G. Chen, Q. Peng, et al., Adv. Mater. 27 (2015) 6195-6201. DOI:10.1002/adma.201502442 |
| [47] |
S. Xu, R. Chen, C. Zheng, et al., Adv. Mater. 28 (2016) 9920-9940. DOI:10.1002/adma.201602604 |
| [48] |
Z. Shuai, Q. Peng, Phys. Rep. 537 (2014) 123. DOI:10.1016/j.physrep.2013.12.002 |
| [49] |
M. Jia, Z. Zhou, M. Lv, et al., Chin. Chem. Lett. 29 (2018) 543-546. DOI:10.1016/j.cclet.2017.09.022 |
| [50] |
E. Lee, S. Gwon, Y. Son, et al., Chin. Chem. Lett. 23 (2012) 484-487. DOI:10.1016/j.cclet.2012.01.020 |
| [51] |
Z. Sun, S. Li, Z. Liu, et al., Chin. Chem. Lett. 27 (2016) 1131-1138. DOI:10.1016/j.cclet.2016.06.007 |
| [52] |
Y. Liu, G. Zhan, Z. Liu, et al., Chin. Chem. Lett. 27 (2016) 1231-1240. DOI:10.1016/j.cclet.2016.06.029 |
| [53] |
H. Wua, B. Wu, X. Yu, et al., Chin. Chem. Lett. 28 (2017) 2151-2154. DOI:10.1016/j.cclet.2017.08.002 |
| [54] |
S. Cai, H. Shi, D. Tian, et al., Adv. Funct. Mater. 28 (2018) 1705045. DOI:10.1002/adfm.201705045 |
| [55] |
P. Xue, P. Wang, P. Chen, et al., Chem. Sci. 8 (2017) 6060-6065. DOI:10.1039/C5SC03739E |
| [56] |
Z. Yang, Z. Mao, X. Zhang, et al., Angew. Chem. Int. Ed. 55 (2016) 2181-2185. DOI:10.1002/anie.201509224 |
| [57] |
L. Bian, H. Shi, X. Wang, et al., J. Am. Chem. Soc. 140 (2018) 10734-10739. DOI:10.1021/jacs.8b03867 |
| [58] |
X. Ma, C. Xu, J. Wang, et al., Angew. Chem. Int. Ed. 57 (2018) 10854-10858. DOI:10.1002/anie.201803947 |