 2019, Vol. 30
2019, Vol. 30 


b Center for Organic Photonics and Electronics Research(OPERA), Kyushu University, Fukuoka 819-0395, Japan;
c School of Physics and Electronics, Shandong Normal University, Ji'nan 250014, China;
d Japan Science and Technology Agency ERATO, Adachi Molecular Exciton Engineering Project, International Institute for Carbon Neutral Energy Research(WPI-IbCNER), Kyushu University, Fukuoka 819-0395, Japan
In the past two decades, immense attention has been received in the area of coordination polymers (CPs) or metal-organic frameworks (MOFs) due to their diverse and remarkable structural related properties, multi-functionality and promising applications in gas storage, separation science, catalysis, sensing, displays, and optical devices [1-8]. In this realm, one of advantages for such functional organic-inorganic hybrid materials is that they can accomplish the synergy of multifunction from inorganic connecting linkers and organic ligands [9, 10]. Compared with luminescent coordination complexes based on noble metals, such as Os(Ⅱ), Ir(Ⅲ) and Pt(Ⅱ), copper(Ⅰ) and silver(Ⅰ) coordination complexes show fascinating photoluminescence properties and potential applications with much lower cost since copper and silver are both abundant elements on the earth [11-22]. For example, copper(Ⅰ)and silver(Ⅰ) complexes with phosphorescence and/or thermally activated delayed fluorescence (TADF) which could be potentially used as highly efficient emitter materials for organic light emitting diodes (OLEDs) [23-26].
Phosphorescent emitter materials can harvest both singlet and triplet excitons due to singlet-triplet state mixing via efficient spin orbit coupling [27, 28]. Therefore phosphors are attracted much interest in both fluorescent lamps and light-emitting diodes. By utilizing Cu(Ⅰ)-based crystalline coordination polymers, J. Li et al. had endeavored to achieve high performance luminescence phosphors [29-32]. Besides phosphors, TADF emitters have been proved to be the promising and practical option for next generation OLEDs in commercial market as well [33-35]. No matter for metalfree TADF materials with donor-acceptor (D-A) motif, or metal complexes for phosphorescence OLEDs (PhOLEDs), shorter excited lifetime will benefit the device performance such as slow efficiency roll-off, long device duration [36-38]. Now the luminescent Cu(Ⅰ) and Ag(Ⅰ) coordination polymers based on D-A organic ligands still need tobe exploited, especiallywith phosphorescence and/or TADF.
In this work, we explored three new copper(Ⅰ) and silver(Ⅰ) coordination polymers based on donor-acceptor motif ligands. The ligands we adopted bear twisting donor acceptor architecture to effectively separate the spatial distribution of HOMO and LUMO, which we supposed would raise the possibility of the accomplishment of small ΔEST, that is the energy difference between lowest excited triplet state (T1) and lowest excited singlet state (S1). Three coordination polymers exhibit similar structural feature with onedimensional (1D) linear chains, as revealed by the single crystals X-ray analysis. The photoluminescence of CP1 and CP2 are both red-shifted with much higher PLQY and longer lifetime, comparing with their corresponding ligands. The temperature-dependent PL decay measurements and PLQY characteristics illustrated that the PL emission of CP1 and CP2 is both fast phosphorescence from 50 to 300 K. Whilst, by introducing Ag(Ⅰ) ion as metal connectors, The photoluminescence of CP3 was proved to possess TADF emission.
The ligands (L1 and L2) of donor-acceptor motif designed in this study are composed of pyridine as acceptor (A), connected to dihydrophenazine and spiroacridine moieties as donors (D) as illustrated in Scheme S1 (Supporting information), which were prepared with a Buchwald-Hartwig amination reaction. All the materials were fully characterized by 1H NMR, 13C NMR and high-resolution mass spectrometry (HRMS). In L1, two parallel terminal pyridine groups are basically vertical to the central dihydrophenazine moiety with the dihedral angles of 89.241(45)° and 89.241(45)° respectively (Fig. S1a in Supporting information). The structure of L2 is presented by Fig. S1b (Supporting information), which revealed the spiroacridine rings are both planar and vertical to each other with the dihedral angle of 89.190 (55)°. And the dihedral angles between two pyridine and neighboring spiroacridine ring are 85.684(68)° and 83.618(103)°, respectively. Above all, in two ligands, the donor segment and acceptor segment adopt a highly twisted configuration in both.
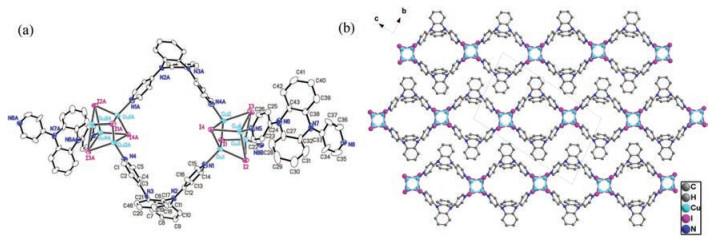
Reactions between ligands and metal salts at room temperature through solvent diffusion method afforded coordination polymers CP1, CP2 and CP3. Single-crystal X-ray diffraction pattern revealed that CP1 crystallizes in the triclinic system P-1. The asymmetry unit consists of four CuI, and two L1 ligand. As shown in Fig. 1, all CuI ion in CP1 are four coordinated (CuI3N) and lie in the center of tetrahedron coordination environment (Cu-I bond lengths of d = 2.664(2)–2.706(2) Å). The Cu4I4 cluster core are formed by four CuI moieties as the connection nodes in CP1 with Cu…Cu distances between 2.602(2)–2.824(2) Å. These Cu4I4 cores lie in a bent coordination environment {Cu4I4N2} which is composed of two terminal pyridyl N-donors with Cu-N bond lengths of 2.01(1) and 2.031(9) Å). Solid-state structure of CP1 exhibits 1D linear chain motif consisting of the boxlike Cu4I4(L1)2 units (Fig. S2 in Supporting information), in which two saddleshaped L1 serve as the linker to bind the adjacent Cu4I4 cluster cores through Cu-N coordination bonds. As shown in Fig. 1, the formed 1D chains are stagger arrangement and extend along the crystallographic [011] direction, in which no obvious interchain interactions are observed.

|
Download:
|
| Fig. 1. (a) ORTEP figure of CP1 (30% probability displacement ellipsoids); (b) Packing pattern (view along a axis) of CP1 (H atoms have been omitted for clarity). | |
{kind=link}
CP2 crystallizes the triclinic system with P-1 space group. The asymmetry unit consists of one CuI, one L1 ligand and one chloroform molecule as guest. CuI ion in CP2 are also four coordinated (CuI2N2) and lie in the center of tetrahedron coordination environment (Cu--I bond length of d = 2.629(1) Å). It is similar to CP1, CP2 also adopts a 1D chain motif in which the Cu2I2 dimer instead of Cu4I4 cluster core acts as the node. The Cu2I2 cluster core are formed by two CuI moieties with Cu…Cu distances of 2.603(1) Å. It is different from CP1, the ligand conformation in boxlike unit of Cu2I2(L2)2 in CP2 is identical to that of free ligand, which is evidenced by the observed dihedral angles between two pyridine and neighboring spiroacridine ring in CP2 are 82.132(214)° and 72.954(111)°, respectively (Fig. S3 in Supporting information). Crystal stacking pattern revealed that there is no intermolecular spatial contacts to be obtained in CP2 (Fig. 2).

|
Download:
|
| Fig. 2. Packing pattern (view along a axis) of CP2 (H atoms and chloroform as solvent molecule have been omitted for clarity). | |
{kind=link}
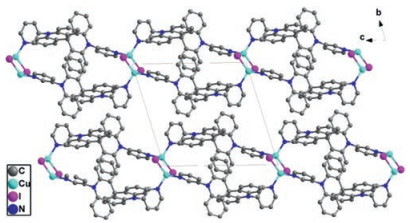
And CP3 crystallized in the monoclinic system with P21/c space group. And the asymmetric unit is composed of one AgI cation, one L2 anion, one CF3CO2- anion and one chloroform molecule. Fig. 3 exhibited that the Ag ion is coordinated by two nitrogen atoms from pyridine terminal groups of two individual L2 and two oxygen atoms from two CF3CO2- anion, presenting a distorted tetrahedron geometry (Ag--N bond length of d = 2.233(7)–2.259(7) Å; Ag--O bond length of d = 2.464(7)–2.471(8) Å). Similar with CP2, the ligand conformation in CP3 is identical to that of L2. The dihedral angles between two pyridine and neighboring spiroacridine ring in CP3 are 86.101° and 76.406°, respectively. With Ag ion acting as connecting node, CP3 adopts a 1D chain in the first place. Furthermore, the adjacent AgI chains can be expanded by CF3CO2- anion to double chains via Ag--O bonds (Fig. 3). It turned out that there is no intermolecular spatial contact between double chains, as presented by crystal stacking pattern (Fig. S4 in Supporting information). The thermogravimetric analysis (TGA) revealed that the decomposition temperatures (Td) with 5% loss of CP1, CP2 and CP3 are all over 320 ℃ (Figs. S5–S7 in Supporting information). And three coordination polymers exhibit homogeneity in solid state as revealed by the agreements between single crystal and as synthesized solid product in powder X-ray diffraction (PXRD) analysis (Figs. S8–S10 in Supporting information).

|
Download:
|
| Fig. 3. (a) ORTEP figure of CP3 (30% probability displacement ellipsoids); (b) Double chain moiety in CP3 (H atoms and chloroform as solvent molecules have been omitted for clarity). | |
{kind=link}
The DFT calculation revealed that the HOMO and LUMO of L1 and L2 are mainly dispersed over the electron-donating moiety and the acceptor moiety, respectively (Fig. S11 in Supporting information) [39]. Because of the big dihedral angles between donor and acceptor units revealed by single crystal analysis, the overlap between the HOMO and LUMO is clearly minimized by the effective separation of their electron densities, frequently implying a small energy difference between the singlet and triplet excited states [33-36]. The ΔEST values of L1 and L2 are calculated to be 0.38 and 0.40 eV, respectively, as illustrated in Table S1 (Supporting information). By cyclicvoltammetry (CV), the first oxidation and reduction potentials were measured in DCM and dimethylformamide (DMF) solutions, respectively. As shown in Fig. S12, two ligands present the similar reduction potential of -2.34 V (for L1) and -2.36 V (for L2) respectively, which can be assigned to the reduction of the pyridine acceptor moieties. The oxidation potentials are -0.20 and 0.55 V, which is in good agreement with the electron-donating capabilities of dihydrophenazine and spiroacridine moiety. Thus, the LUMO energy levels of L1 and L2 were estimated to be -2.00 and -1.97 eV, and the HOMO energy levels as -4.32 and -5.37 eV, respectively (Table S2 in Supporting information) [40].
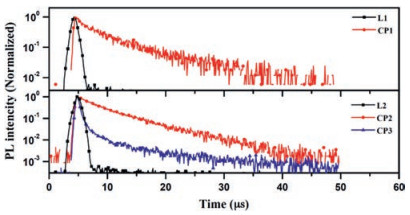
As presented in Fig. S13 (Supporting information), ligand L1 exhibit absorption bands in the region of 350 nm to 420 nm, with moderately weak molar absorption coefficient ε, which can be ascribed to the intramolecular charge-transfer (ICT) transition from the HOMO to the LUMO. Whilst, the absorption spectra of L2 do not present observable ICT states in the longer-wavelength region, indicating quite weak CT interaction in its ground states. Nevertheless, the photoluminescence (PL) spectra of both ligands exhibit broad and structureless emission, indicating that their emissive S1 excited states are ICT states in nature. Further, the emission maxima of L2 are blue-shifted relative to that of L1, corresponding to their lower electron-donating capabilities. Meanwhile, the phosphorescence spectra of them captured at 77 K are all well resolved and show characteristic vibrational structures, indicating that their lowest emissive T1 excited states are 3LE at this temperature [33-36]. Based on the values calculated from their fluorescence and phosphorescence spectra, the ΔEST of L1 and L2 are estimated to be 0.45 and 0.50 eV, respectively (Table S3 in Supporting information). L1 and L2 in solid state exhibit sky blue and blue ICT emission with the maximum wavelength of 501 nm and 412 nm, as illustrated in Figs. S14 and S15 (Supporting information). Unlike their TADF analogues [41, 42], Fig. 4 revealed that the transient PL decay characteristics of two ligands, with pyridine as weaker acceptor, both exhibits one component decay only, which means the big barriers from T1 to S1 would definitely hinder the occurrence of TADF process for both ligands. In comparison with two ligands, coordination polymer CP1 and CP2 both exhibited red-shift of PL emission and higher PLQY of 15% and 46% respectively (Table S4 and Fig. S15). The temperature-dependent transient PL decay measurements (Fig. S17 in Supporting information) illustrate that the luminescence lifetimes at 75 K of both coordination polymers (CP1 and CP2) are 12 and 11 μs respectively. As shown in Fig. S17, the PL emission life times of CP1 and CP2 both were revealed to have little variation with 5–12 μs in the temperature range of 75–300 K, and the emission spectra remain unchanged despite the variation of temperature, which indicate not effective reverse intersystem crossing (RISC) but spin-orbit coupling (SOC) to realized radiative transition from first excited triplet state to the ground state. Meanwhile, the spin-orbit coupling matrix elements of CP1 and CP2 were calculated based on the quadratic response function method by Dalton package [43, 44]. Small SOC effect (1.44 cm-1 for CP1 and 1.52 cm-1 for CP2) show that the ISC process from S1 to T1 is preferably occurred. The characteristics of temperature-dependent PLQYs of CP1 and CP2 prove the absence of TADF, since the PLQYs of both CP1 and CP2 are actually reduced with the increase of temperature, which is the typical behavior of triplet emitter (Table S5 in Supporting information). To better understand the observation, the HOMO and LUMO of CP1 and CP2 were calculated using density functional theory (DFT) with the Gaussian 09 package at the B3LYP/6-31G(d) level [39], which turned out to be mainly dispersed over rather copper cluster than ligands themselves (Fig. S19 in Supporting information).

|
Download:
|
| Fig. 4. Transient PL decay spectra at 300 K of L1, L2, CP1, CP2 and CP3 in solid state. | |
{kind=link}
Different from the reported TADF Cu(Ⅰ) complexes [24-26], CuI cluster-based coordination complexes usually exhibit phosphorescence due to effective intersystem crossing (ISC) [30-32]. In order to realize TADF property, Ag(Ⅰ) ion was applied for constructing coordination polymer instead of CuI. The DFT calculation illustrated that the HOMO and LUMO of CP3 are mainly dispersed over the electron-donating moiety and the acceptor moiety, respectively (Fig. S20 in Supporting information). And the overlap between the HOMO and LUMO of CP3 is clearly minimized by the effective separation of their electron densities due to large dihedral angles between donor and acceptor units, which is in good agreement with single crystal analysis as well. The difference of the electron configuration between CP1, CP2 and CP3 may result in different PL emission nature of them. Comparing with CP2 derived from the same ligand, CP3 exhibited similar PL emission with small blue-shift in spectra, as indicated in Fig. S16 (Supporting information). Comparing with L2, higher PLQY with 34% of CP3 suggest non-radiative decay via intramolecular rotation and vibrational motions could be suppressed in coordination polymers [45]. The temperature-dependent transient PL decay measurements revealed that CP3 clearly exhibited sky blue emission with both the prompt and delayed components (Fig. 4). The lifetime of the delayed component at room temperature is 7.7 μs. The increase of PL emission intensity with temperature is consistent with TADF materials (Fig. S18a and Table S5 in Supporting information). The delayed emission spectrum of CP3 is slightly red-shifted from that of the prompt emission (Fig. S18b in Supporting information), which is usual for TADF materials and could be explained by structural relaxation [33-38].
In this communication, we have investigated the synthesis of three luminescent Cu(Ⅰ) and Ag(Ⅰ) coordination polymers. Two Cu(Ⅰ) coordination polymers, CP1 and CP2, exhibit PL emission with much higher PLQY and longer lifetime, comparing with those of the corresponding ligands L1 and L2. Unfortunately, TADF has not been observed in both coordination polymers due to effective ISC process with the involvements of copper iodine clusters as metal connections. By applying Ag(Ⅰ) cation as metal connectors, the resulting coordination polymers, CP3, realize sky blue PL emission with TADF component.
AcknowledgmentsThis work was financially supported by the National Natural Science Foundation (Nos. 21772116, 21671122 and 21475078), the Shandong Taishan Scholar’s Construction Project, JSPS KAKENHI (No. JP17H01232) and the Japan Science and Technology Agency (JST), ERATO, Adachi Molecular Exciton Engineering Project, under JST ERATO (No. JPMJER1305), Japan.
Appendix A. Supplementary dataSupplementary material related to this article canbefound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.08.006.
| [1] |
H.C. Zhou, J.R. Long, O.M. Yaghi, Chem. Rev. 112 (2012) 673-674. DOI:10.1021/cr300014x |
| [2] |
L.E. Kreno, K. Leong, O.K. Farha, et al., Chem. Rev. 112 (2012) 1105-1125. DOI:10.1021/cr200324t |
| [3] |
Y. Cui, Y. Yue, G. Qian, B. Chen, Chem. Rev. 112 (2012) 1126-1162. DOI:10.1021/cr200101d |
| [4] |
M.P. Suh, H.J. Park, T.K. Prasad, D.W. Lim, Chem. Rev. 112 (2012) 782-835. DOI:10.1021/cr200274s |
| [5] |
J.R. Li, J. Sculley, H.C. Zhou, Chem. Rev. 112 (2012) 869-932. DOI:10.1021/cr200190s |
| [6] |
Z. Hu, B.J. Deibert, J. Li, Chem. Soc. Rev. 43 (2014) 5815-5840. DOI:10.1039/C4CS00010B |
| [7] |
J.L. Wang, C. Wang, W. Lin, ACS Catal. 2 (2012) 2630-2640. DOI:10.1021/cs3005874 |
| [8] |
X.J. Wei, Y.H. Li, Z.B. Qin, G.H. Cui, J. Mol. Struct. 1175 (2019) 253-260. DOI:10.1016/j.molstruc.2018.08.001 |
| [9] |
Y. Cui, B. Li, H. He, et al., Acc. Chem. Res. 49 (2016) 483-493. DOI:10.1021/acs.accounts.5b00530 |
| [10] |
N.X. Zhu, C.W. Zhao, J.C. Wang, Y.A. Li, Y.B. Dong, Chem. Commun. 52 (2016) 12702-12705. DOI:10.1039/C6CC07027B |
| [11] |
P.C. Ford, E. Cariati, J. Bourassa, Chem. Rev. 99 (1999) 3625-3648. DOI:10.1021/cr960109i |
| [12] |
R. Peng, M. Li, D. Li, Coord. Chem. Rev. 254 (2010) 1-18. DOI:10.1016/j.ccr.2009.10.003 |
| [13] |
Z.W. Liu, M.F. Qayyum, C. Wu, et al., J. Am. Chem. Soc. 133 (2011) 3700-3703. DOI:10.1021/ja1065653 |
| [14] |
T. Hasegawa, A. Kobayashi, H. Ohara, M. Yoshida, M. Kato, Inorg. Chem. 56 (2017) 4928-4936. DOI:10.1021/acs.inorgchem.6b03122 |
| [15] |
J. Nitsch, F. Lacemon, A. Lorbach, et al., Chem. Commun. 52 (2016) 2932-2935. DOI:10.1039/C5CC09659F |
| [16] |
S.Z. Zhan, M. Li, J. Zheng, et al., Inorg. Chem. 56 (2017) 13446-13455. DOI:10.1021/acs.inorgchem.7b02144 |
| [17] |
M.Z. Shafikov, A.F. Suleymanova, R. Czerwieniec, H. Yersin, Chem. Mater. 29 (2017) 1708-1715. DOI:10.1021/acs.chemmater.6b05175 |
| [18] |
M.Z. Shafikov, A.F. Suleymanova, A. Schinabeck, H. Yersin, J. Phys. Chem. Lett. 9 (2018) 702-709. DOI:10.1021/acs.jpclett.7b03160 |
| [19] |
A. Kobayashi, T. Hasegawa, M. Yoshida, M. Kato, Inorg. Chem. 55 (2016) 1978-1985. DOI:10.1021/acs.inorgchem.5b02160 |
| [20] |
B. Huitorel, H.E. Moll, M. Cordier, et al., Inorg. Chem. 56 (2017) 12379-12388. DOI:10.1021/acs.inorgchem.7b01870 |
| [21] |
B. Hupp, J. Nitsch, T. Schmitt, et al., Angew. Chem. Int. Ed. 57 (2018) 13671-13675. DOI:10.1002/anie.201807768 |
| [22] |
S. Evariste, A.M. Khalil, M.E. Moussa, et al., J. Am. Chem. Soc. 140 (2018) 12521-12526. DOI:10.1021/jacs.8b06901 |
| [23] |
M.J. Leitl, V.A. Krylova, P.I. Djurovich, M.E. Thompson, H. Yersin, J. Am. Chem. Soc. 136 (2014) 16032-16038. DOI:10.1021/ja508155x |
| [24] |
T. Hofbeck, U. Monkowius, H. Yersin, J. Am. Chem. Soc. 137 (2015) 399-404. DOI:10.1021/ja5109672 |
| [25] |
X.L. Chen, R. Yu, X.Y. Wu, D. Liang, J.H. Jia, C.Z. Lu, Chem. Commun. 52 (2016) 6288-6291. DOI:10.1039/C6CC00809G |
| [26] |
A. Schinabeck, M.J. Leitl, H. Yersin, J. Phys. Chem. Lett. 9 (2018) 2848-2856. DOI:10.1021/acs.jpclett.8b00957 |
| [27] |
M.A. Baldo, D.F. O'Brien, Y. You, et al., Nature 395 (1998) 151-154. DOI:10.1038/25954 |
| [28] |
C. Adachi, M.A. Baldo, M.E. Thompson, S.R. Forrest, J. Appl. Phys. 90 (2001) 5048-5051. DOI:10.1063/1.1409582 |
| [29] |
X. Zhang, W. Liu, G.Z. Wei, et al., J. Am. Chem. Soc. 136 (2014) 14230-14236. DOI:10.1021/ja507927a |
| [30] |
Q. Gong, Z. Hu, B.J. Deibert, et al., J. Am. Chem. Soc. 136 (2014) 16724-16727. DOI:10.1021/ja509446h |
| [31] |
W. Liu, Y. Fang, G.Z. Wei, et al., J. Am. Chem. Soc. 137 (2015) 9400-9408. DOI:10.1021/jacs.5b04840 |
| [32] |
Y. Fang, W. Liu, S.J. Teat, et al., Adv. Funct. Mater. 27 (2017) 1603444. DOI:10.1002/adfm.201603444 |
| [33] |
H. Uoyama, K. Goushi, K. Shizu, H. Nomura, C. Adachi, Nature 492 (2012) 234-238. DOI:10.1038/nature11687 |
| [34] |
Q. Zhang, B. Li, S. Huang, et al., Nat. Photonics 8 (2014) 326-332. DOI:10.1038/nphoton.2014.12 |
| [35] |
M.Y. Wong, E. Zysman-Colman, Adv. Mater. 29 (2017) 1605444. DOI:10.1002/adma.201605444 |
| [36] |
Y. Geng, A. D'Aleo, K. Inada, et al., Angew. Chem. Int. Ed. 56 (2017) 16536-16540. DOI:10.1002/anie.201708876 |
| [37] |
A. Yusoff, A.J. Huckaba, M.K. Nazeeruddin, Top. Curr. Chem. 375 (2017) 39. DOI:10.1007/s41061-017-0126-7 |
| [38] |
T.L. Wu, M.J. Huang, C.C. Lin, et al., Nat. Photonics 12 (2018) 235-240. DOI:10.1038/s41566-018-0112-9 |
| [39] |
M.J. Frisch, et al., Gaussian09, revision E.01, Gaussian, Inc., Wallingford, CT, 2009.
|
| [40] |
P.I. Djurovich, E.I. Mayo, S.R. Forrest, M.E. Thompson, Org. Electron. 10 (2009) 515-520. DOI:10.1016/j.orgel.2008.12.011 |
| [41] |
J. Lee, K. Shizu, H. Tanaka, et al., J. Mater. Chem. C 3 (2015) 2175-2181. DOI:10.1039/C4TC02530J |
| [42] |
M. Liu, R. Komatsu, X. Cai, et al., Chem. Mater. 29 (2017) 8630-8636. DOI:10.1021/acs.chemmater.7b02403 |
| [43] |
DALTON. A molecular electronic structure program, http://daltonprogram.org/
|
| [44] |
Q. Peng, D. Fan, R. Duan, et al., J. Phys. Chem. C 121 (2017) 13448-13456. DOI:10.1021/acs.jpcc.7b00692 |
| [45] |
Z. Wei, Z.Y. Gu, R.K. Arvapally, et al., J. Am. Chem. Soc. 136 (2014) 8269-8276. DOI:10.1021/ja5006866 |