 2019, Vol. 30
2019, Vol. 30 


Preorganization is one of the key concepts in supramolecular chemistry field [1]. The common approach toward preorganization involves the fabrication of macrocyclic and molecular tweezer-like receptors, which limit the conformational degrees of freedom available [2, 3]. These rigid structures provide definite size and constrained environment for guest accommodation. For the resulting host-guest complexation system, a variety of noncovalent bonds can be incorporated in a multivalent and cooperative manner [4]. In this respect, we and others have constructed molecular tweezers and macrocycles with the presence of square-planar terpyridine platinum(Ⅱ) units [5-7]. Thanks to the preorganization effect, these supramolecular receptors provide synergistic metal–metal and donor–acceptor interactions toward the complementary organometallic guests. The resulting host-guest systems are endowed with fascinating photo-physics and stimuli responsiveness, which are promising for biomedical and photo-catalytic applications [5d, 6f, 6i]. Despite the great progress achieved so far, it still requires deeper understanding of the preorganization effect, which facilitates to evaluate the contribution of non-covalent forces in a precise and quantitative manner.
Herein we have shown an interesting example, in which remarkable discrepancy exists between macrocyclic and acyclic preorganization modes [8]. Specifically, two structurally similar receptors 1-2 (Scheme 1) have been designed. Macrocycle 1 preserves the basic structural feature of molecular tweezer 2, while an extra C10 alkyl linker is tethered between two terpyridine platinum(Ⅱ) [Pt(Ⅱ)(N^N^N)(C≡C-R)] units to achieve macrocyclization. Both 1 and 2 are capable of encapsulating neutral organoplatinum(Ⅱ) [Pt(Ⅱ)(C^N^C)(C≡N-R)] guest 3 (Scheme 1). Pt(Ⅱ)…Pt(Ⅱ) metal–metal forces are prone to form, because of the proximity of Pt(Ⅱ) atoms between host and guest species. Unexpectedly, the subtle structural difference between 1 and 2 gives rise to huge difference on the number of Pt(Ⅱ)…Pt(Ⅱ) metal– metal bonds. In particular, two-fold Pt(Ⅱ)…Pt(Ⅱ) bonds are involved in macrocycle/guest complex 1/3, while only one-fold bond exists for molecular tweezer/guest complex 2/3. The optical properties, non-covalent binding strength, and photosensitization capability of the host-guest complexes can be further influenced by these two preorganization modes.

|
Download:
|
| Scheme 1. Schematic representation for the host‒guest complexes 1/3 and 2/3, by employing structurally similar macrocycle and molecular tweezer as the synthetic receptors. Triflate is served as the counterion in both 1 and 2, which are omitted in the chemical structures. | |
{kind=link}
The designed macrocycle 1 and molecular tweezer 2 are synthesized quite straightforward (Schemes S1 and S2 in Supporting information). Both synthetic routes involve copper– catalyzed platinum–carbon coupling reactions as the final step. The identity and purity of 1-2 and the synthetic intermediates are unambiguously confirmed by means of 1H, 13C NMR and ESI–MS spectra (Figs. S11–S25 in Supporting information). For 1 in chloroform, two absorbance bands are observed in the visible region (λmax =447 nm and 472 nm, Fig. 1a). According to the previous literatures, they are assigned to MLCT/LLCT (metal-toligand and ligand-to-ligand charge transfer) transition of the [Pt(Ⅱ)(N^N^N)(C≡C-R)] units [5a]. Moreover, MLCT/LLCT emission band of 1 is centred at 582 nm, together with a shoulder band at 620 nm (Fig. 1b). The large Stokes shift, along with the long lifetime in microsecond range (τ =1.57 μs, Table S1 in Supporting information), illustrate the triplet origin of the MLCT/LLCT emission signals.

|
Download:
|
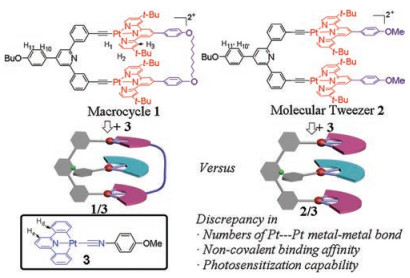
| Fig. 1. (a) UV–vis spectra of 1 (black line) and 2 (red line) (c = 0.25 mmol/L in CHCl3). (b) Emission spectra of 1 (black line) and 2 (red line) (c = 0.10 mmol/L in CHCl3, λex = 440 nm). Inset of (b): images of 1 and 2 under 365 nm UV lamp. Optimized geometries of (c) 1, and (d) 2 via DFT calculation. Pt atoms are described by Lanl2dz, while the residual atoms are described by PBEPBE./3-21G. | |
{kind=link}
Remarkably, the MLCT/LLCT signals of 1 are bathochromic-shift than those of 2 (absorbance: λmax = 425 nm and 464 nm, emission: λmax = 567 nm, Figs. 1a and b). In view of the structural similarity between 1 and 2, their distinct MLCT/LLCT spectroscopic signals are elucidated by means of DFT (density functional theory) calculations. For the optimized geometry of 1 (Fig. 1c) [9], the two [Pt(Ⅱ)(N^N^N)(C≡C-R)] pincers are almost coplanar to each other, giving rise to the short Pt(Ⅱ)…Pt(Ⅱ) distance of 6.6 Å. In comparison, the two pincers on 2 are more twisted to form a Vshape conformation, accompanied by the long Pt(Ⅱ)…Pt(Ⅱ) distance of 10.9 Å (Fig. 1d). Meanwhile, the dihedral angle of the diphenylpyridine spacer varies from 15.5° for 1 to 30.7° for 2. Hence, 1 possesses higher effective π-conjugation length than that of 2, contributing to the bathochromic shifts of MLCT/LLCT signals.
Non-covalent complexation behaviors are further studied between the positively-charged receptors 1-2 and neutral guest 3. For 3 itself, the intra-ligand (IL) absorption is located at 320– 400 nm, while no emission signal can be observed (Fig. S1 in Supporting information) [10]. Upon mixing an equimolar amount of 1 and 3 together in CHCl3, a low-energy absorbance appears between 520 nm and 640 nm (Fig. 2a), which is absent for the individual species. With reference to the previous reports [5a], the emergent band is characteristic for Pt(Ⅱ)…Pt(Ⅱ) MMLCT (metalmetal-to-ligand charge transfer) transitions. The phenomenon suggests the formation of non-covalent host-guest complex, leading to the proximity of heterologous Pt atoms between 1 and 3. Although the MMLCT absorbance exists upon mixing 2 and 3 together (ranging from 520 nm to 620 nm, Fig. 2a), the extinction coefficient value of 2/3 is 3 times lower than that of 1/3 (at 550 nm, ε: 1.91 ×103 L mol-1 cm-1 of 1/3, versus 0.65 ×103 L mol-1 cm-1 of 2/3).

|
Download:
|
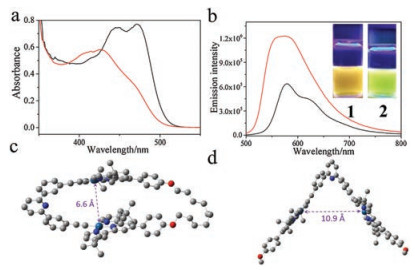
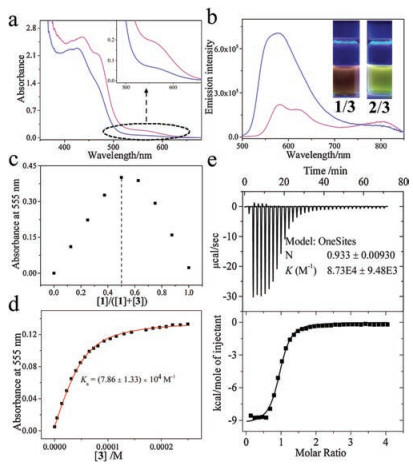
| Fig. 2. (a) Absorption, and (b) emission spectra of 1/3 (pink lines) and 2/3 (blue lines) (c = 0.10 mmol/L for each compound in CHCl3). Inset of (b): images of complexes 1/3 and 2/3 under 365 nm UV lamp. (c) Job's plot of 1/3. (d) Intensity changes of MMLCT absorbance upon titrating 3 into the chloroform solution of 1 (0.05 mmol/L in CHCl3). The red line denotes non-linear mathematical fitting. (e) ITC data by titrating 3 (8.00 mmol/L in CHCl3, 298 K) into the chloroform solution of 1 (0.40 mmol/L in CHCl3). | |
{kind=link}
Moreover, the two host-guest complexes exhibit distinct emission properties (Fig. 2b). In particular, a near infrared MMLCT emission signal appears at 803 nm for 1/3, accompanying by the severe quenching of MLCT/LLCT emission signal. On the contrary, the MMLCT emission intensity of 2/3 is significantly lower than that of the MLCT/LLCT signal derived from 2. As a consequence, brown and green emission colours are visualized under 365 nm UV lamp for complexes 1/3 and 2/3, respectively (Fig. 2b, inset).
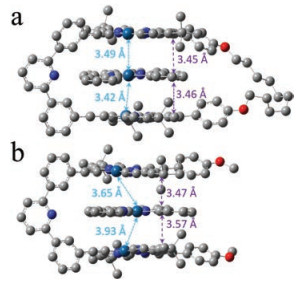
To explain the intensive MMLCT signals in complex 1/3 than that of 2/3, DFT theoretical calculations are further employed. As shown in Figs. 3a and b, guest 3 is encapsulated into the cavities of the host receptors for both complexes [9]. In terms of 1/3, the interplanar distances between 3 and two [Pt(Ⅱ)(N^N^N)(C≡C-R)] pincers on macrocycle 1 are calculated to be 3.45 Å and 3.46 Å, respectively (Fig. 3a). Simultaneously, intermolecular Pt(Ⅱ)…Pt(Ⅱ) distances are determined to be 3.49 Å and 3.42 Å, with the Pt–Pt–Pt angle of 172.6°. Hence, it is evident that both donor-acceptor and Pt(Ⅱ)…Pt(Ⅱ) metal–metal interactions exist in a two-fold manner.For molecular tweezer/guest complex 2/3, it also possesses twofold donor-acceptor forces, as manifested by the interplanar distances of 3.57 Å and 3.47 Å. Nevertheless, the Pt(Ⅱ)…Pt(Ⅱ) distances are calculated to be 3.93 Å and 3.65 Å. The former one is too long to form Pt(Ⅱ)…Pt(Ⅱ) metal–metal bond. As a result, only one-fold metal-metal bond exists for 2/3. Hence, it is evident that the number of Pt(Ⅱ)…Pt(Ⅱ) metal–metal bonds varies for the two preorganization modes.

|
Download:
|
| Fig. 3. Optimized structures of (a) complex 1/3, and (b) complex 2/3 via DFT calculation. Pt atoms are described by Lanl2dz, while the residual atoms are described by PBEPBE./3-21 G. | |
{kind=link}
Moreover, the quantitative binding thermodynamics are compared between 1/3 and 2/3. For both complexes, the binding stoichiometries are determined to be 1:1, as manifested by the Job's plots on the basis of MMLCT absorbance intensity (Fig. 2c and Fig. S6 in Supporting information). The collected MMLCT absorbance at 555 nm is further fitted with one-site model (Eq. S1 in Supporting information), providing Ka value of (7.86 ± 1.33) × 104 L/mol for complex 1/3 (Fig. 2d). The value is determined to be (8.93 ± 0.19) × 104 L/mol on the basis of emission titration experiments (Fig. S3 in Supporting information). Besides, isothermal titration calorimetry (ITC) experiment provides an alternative method to quantify non-covalent binding affinity. Depending on the exothermic isotherm curve, Ka value of 1/3 is determined to be (8.73 ± 0.95) × 104 L/mol (Fig. 2e), which is consistent with the above spectroscopic titration results. Noteworthy, the Ka values of 1/3 are 16- and 9.4-fold higher than those of 2/3 (Ka, UV = (4.93 ± 0.03) × 103 L/mol and Ka, FL = (9.52 ± 0.01) × 103 L/mol, Figs. S4 and S5 in Supporting information) on the basis of absorption and emission measurements, respectively.
The reinforced host-guest complexation strength from acyclic to macrocyclic preorganization are further validated via 1H NMR experiments. For complex 1/3, protons H10, 11 remain almost intact, while the terpyridine protons on 1 and the diphenylpyridine protons on 3 undergo enormously upfield shifts (-0.79, -0.61, -0.71, -1.00 and -0.94 ppm for H1, H2, H3, Hd, and He, respectively, Figs. S7a-c in Supporting information). It supports the presence of donor - acceptor interactions between the positively-charged [Pt (Ⅱ)(N^N^N)(C≡C-R)] units on 1 and the neutral [Pt(Ⅱ)(C^N^C) (C≡N-R)] unit on 3. In comparison, complex 2/3 displays weaker host-guest complexation tendency, as reflected by the less upfield resonances shifts (Δδ = -0.38 and -0.46 ppm for Hd-e, respectively, Figs. S7c-e in Supporting information).
As previously documented, the presence of long-lived triplet excited states renders excellent photosensitization capability to terpyridine platinum(Ⅱ) complexes [11]. Upon formation of host- guest complexes 1/3 and 2/3, the MMLCT transition signals emerge at the low-energy region, which facilitate photosensitization under the mild visible light conditions (Fig. 4a). In this regard, an OLED lamp (12 W, 590 nm) is employed, which overlaps with the MMLCT absorbance of host-guest complexes. Upon photo-irradiating 1/3 (0.05 mmol/L) and 9, 10-dimethylanthracene (DMA, 0.25 mmol/L) together in chloroform, the typical absorbance of DMA (λmax = 360, 380 and 410 nm, Fig. 4b) decline for their intensities. The results suggest energy transfer between the triplet excited state of 1/3 and surrounding molecular oxygen (Fig. 4a). Singlet oxygen (1O2) formed in situ is further captured by DMA to form 9, 10- dimethylanthracene 9, 10-endoperoxide, as verified by upfield shifting of the DMA methyl resonances from 3.22 ppm to 2.26 ppm (Fig. S9 in Supporting information).

|
Download:
|
Fig. 4. (a) Schematic representation for the visible-light photosensitization process (ISC: intersystem crossing; ET: energy transfer). (b) UV–vis absorption changes of 9, 10-dimethylanthracene (0.25 mmol/L in chloroform) upon light irradiation (590 nm, 12 W). Complex 1/3 (c = 0.05 mmol/L) is employed as the photosensitizer. (c) 1O2 generation efficiency of 1 (    |
|
{kind=link}
The quantitative 1O2 generation rate can be further acquired (Eq. S2 in Supporting information), which is 3461 L mol-1 min-1 for complex 1/3 (0.05 mmol/L for each compound, Fig. 4c). The value is significantly higher than those of the individual species (1: 171.4 L mol-1 min-1, 3: 4.10 L mol-1 min-1, Fig. 4c and Fig. S10 in Supporting information). It is highly plausible, since the emergent MMLCT band is crucial for 1O2 production under the visible light irradiation conditions. More intriguingly, 1O2 generation capability of 1/3 is approximately 3-fold higher than that of 2/3 (1209 L mol-1 min-1). Considering that both 1/3 and 2/3 are in dynamic equilibrium between complexed (the real photosensitization species) and uncomplexed states, the different photosensitization capabilities for 1/3 and 2/3 are rationalized via the mathematical calculation (Eq. S3 in Supporting information). In detail, 61% of the complexed species exist for 1/3 at the monomer concentration of 0.05 mmol/L. In stark contrast, only 19% of 2/3 exists in the "active" complexed form. The results unambiguously support that, in addition to the emergent MMLCT absorbance, host-guest binding strength also exerts crucial impact on visiblelight photosensitization efficiency.
In summary, subtle structural variation between macrocycle 1 and molecular tweezer 2 gives rise to the remarkable discrepancy in host-guest complexation behaviours. Although both donor- acceptor and Pt(Ⅱ)…Pt(Ⅱ) metal–metal interactions are involved in both 1/3 and 2/3, these two complexes possess different numbers of Pt(Ⅱ)…Pt(Ⅱ) metal–metal bonds. As a result, 1/3 displays one order of magnitude higher for the non-covalent binding affinity, and 3-fold enhancement for the photosensitized 1O2 generation capability than those of 2/3. The prominent role of preorganization modes (macrocyclic versus acyclic) exemplified in the current work would benefit for the rational design of host- guest systems in future study.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (Nos. 21674106 and 21871245), CAS Youth Innovation Promotion Association (No. 2015365), and the Fundamental Research Funds for the Central Universities (No. WK3450000004).
Appendix A. Supplementary dataSupplementary material related to this article canbefound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.05.007.
| [1] |
D.J. Cram, Angew. Chem. Int. Ed. 25 (1986) 1039-1057. DOI:10.1002/anie.198610393 |
| [2] |
(a) J.M. Lehn, Science 260 (1993) 1762-1763; (b) F. Wang, J. Zhang, X. Ding, et al., Angew. Chem. Int. Ed. 49 (2010) 1090-1094; (c) M. Xue, Y. Yang, X. Chi, X. Yan, F. Huang, Chem. Rev. 115 (2015) 7398-7501; (d) Z. Liu, S.K.M. Nalluri, J.F. Stoddart, Chem. Soc. Rev. 46 (2017) 2459-2478; (e) K. Kotturi, E. Masson, Chem. -Eur. J. 24 (2018) 8670-8678; (f) S. Kuang, Z. Hu, H. Zhang, et al., Chem. Commun. 54 (2018) 2169-2172. |
| [3] |
(a) C.W. Chen, H.W. Whitlock, J. Am. Chem. Soc. 100 (1978) 4921-4922; (b) S.C. Zimmerman, W. Wu, J. Am. Chem. Soc. 111 (1989) 8054-8055; (c) M. Hardouin-Lerouge, P. Hudhomme, M. Salle, Chem. Soc. Rev. 40 (2011) 30-43; (d) C. Shao, M. Stolte, F. Wuerthner, Angew. Chem. Int. Ed. 52 (2013) 7482-7486; (e) S. Ibáñez, M. Poyatos, E. Peris, Angew. Chem. Int. Ed. 56 (2017) 9786-9790; (f) Y. Han, Y. Tian, Z. Li, F. Wang, Chem. Soc. Rev. 47 (2018) 5165-5176; (g) S. Ibáñez, M. Poyatos, E. Peris, Angew. Chem. Int. Ed. 57 (2018) 16816-16820. |
| [4] |
(a) J.D. Badjic, A. Nelson, S.J. Cantrill, W.B. Turnbull, J.F. Stoddart, Acc. Chem. Res. 38 (2005) 723-732; (b) C.A. Hunter, H.L. Anderson, Angew. Chem. Int. Ed. 48 (2009) 7488-7499; (c) L.K.S. von Krbek, C.A. Schalley, P. Thordarson, Chem. Soc. Rev. 46 (2017) 2622-2637. |
| [5] |
(a) Y. Tanaka, K.M.C. Wong, V.W.W. Yam, Chem. Sci. 3 (2012) 1185-1191; (b) Y. Tanaka, K.M.C. Wong, V.W.W. Yam, Angew. Chem. Int. Ed. 52 (2013) 14117-14120; (c) Y. Tanaka, K.M.C. Wong, V.W.W. Yam, Chem. -Eur. J. 19 (2013) 390-399; (d) A.K.W. Chan, W.H. Lam, Y. Tanaka, K.M.C. Wong, V.W.W. Yam, Proc. Natl. Acad. Sci. U. S. A. 112 (2015) 690-695; (e) F.K.W. Kong, A.K.W. Chan, M. Ng, K.H. Low, V.W.W. Yam, Angew. Chem. Int. Ed. 56 (2017) 15103-15107. |
| [6] |
(a) Y.K. Tian, Y.G. Shi, Z.S. Yang, F. Wang, Angew. Chem. Int. Ed. 53 (2014) 6090-6094; (b) Y.K. Tian, Y.F. Yang, Z.S. Yang, F. Wang, Macromolecules 49 (2016) 6455-6461; (c) Z. Gao, Y. Han, J. Chen, X. Wang, F. Wang, Chem. -Asian. J. 11 (2016) 1775-1779; (d) X. Lv, Y. Han, Z. Yang, et al., Tetrahedron Lett. 57 (2016) 1971-1975; (e) Z. Li, Y. Han, F. Jin, et al., Daltron Trans. 45 (2016) 17290-17295; (f) Z. Li, Y. Han, Z. Gao, F. Wang, ACS Catal. 7 (2017) 4676-4681; (g) T. Fu, Z. Li, Z. Zhang, X. Zhang, F. Wang, Macromolecules 50 (2017) 7517-7525; (h) Z.G.Y. Han, S. Chen, et al., ACS Macro Lett. 6 (2017) 541-545; (i) Z. Li, Y. Han, Z. Gao, T. Fu, F. Wang, Mater. Chem. Front. 2 (2018) 76-80; (j) X. Zhang, L. Ao, Y. Han, Z. Gao, F. Wang, Chem. Commun. 54 (2018) 1754-1757; (k) Z. Gao, Y. Han, Z. Gao, F. Wang, Acc. Chem. Res. 51 (2018) 2719-2729. |
| [7] |
(a) A.J. Goshe, I.M. Steele, B. Bosnich, J. Am. Chem. Soc. 125 (2003) 4444-4450; (b) J.D. Crowley, I.M. Steele, B. Bosnich, Inorg. Chem. 44 (2005) 2989-2991; (c) Y. Yamaki, T. Nakamura, S. Suzuki, et al., Eur. J. Org. Chem. (2016) 1678-1683. |
| [8] |
(a) D. Canevet, M. Gallego, H. Isla, et al., J. Am. Chem. Soc. 133 (2011) 3184-3190; (b) J.M. McGrath, M.D. Pluth, J. Org. Chem. 79 (2014) 711-719. |
| [9] |
To reduce the computational costs, the peripheral 4-butoxyphenyl groups on both 1 and 2 are removed during DFT calculation processes.
|
| [10] |
W. Lu, M.C. Chan, K.K. Cheung, C.M. Che, Organometallics 20 (2001) 2477-2486. DOI:10.1021/om0009839 |
| [11] |
(a) D. Zhang, L.Z. Wu, L. Zhou, et al., J. Am. Chem. Soc. 126 (2004) 3440-3441; (b) Q.Y. Meng, T. Lei, L.M. Zhao, et al., Org. Lett. 16 (2014) 5968-5971. |