 2019, Vol. 30
2019, Vol. 30 


b CAS Center for Excellence in Nanoscience, CAS Key Laboratory of Nanosystem and Hierarchical Fabrication, National Center for Nanoscience and Technology(NCNST), Beijing 100190, China
Photon upconversion (UC), an anti-Stokes type emission which could absorb two or more low-energy photons leading to the emission of light with higher energy level, has attracted enormous attentions in a wide range of disciplines in the last few years [1, 2]. Among the various upconversion systems, triplet-triplet annihilation-based photon upconversion (TTA-UC) involves bimolecular process following the Dexter energy transfer mechanism [3-5] is particularly useful, which has been widely studied in various fields. The low excitation power density (as low as 0.1 mW/cm2) [6-9] render TTA-UC the ability to be used in photovoltaics, artificial photosynthesis, photocatalysis and optics, etc. [9-17]. Generally, the efficient TTA-UC could be observed in low viscous solutions, as it allows fast diffusion and collision of excited molecules. However, from the viewpoint of practical application, TTA-UC in liquid state is limited for wide applications [18-21], which makes the TTA-UC system in rigid materials imperative to be developed [22, 23]. However, great difficulties have been encountered in the process of building TTA-UC in rigid hosts, such as crystal and polymer for the good mobility is essential for the triplet donor or acceptor molecules in the processes of triplet-triplet energy transfer (TTET) and TTA. Therefore, it is not easy for TTA-UC system to be applied to rigid device [24-26]. As a compromise way, supramolecular selfassembly provides one powerful solution to achieve highly efficient UC in quasi-solid state, such as the supramolecular gels [18, 27-33], in which the donor and acceptor could be organized in a well-ordered arrangement, enabling the highly efficient TTA-UC by enhancing the efficiency of TTET and triplet energy migration among the highly organized chromophores.
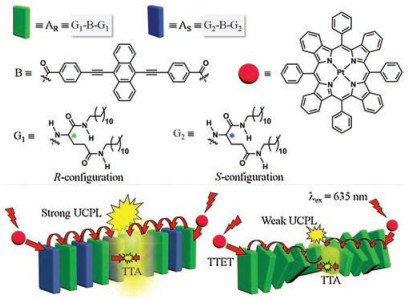
In most of the supramolecular self-assemble processes, chirality could be one significant factor in the aspect of controlling the arrangement of the gelators or obtaining expected structure of assemblies. In line with this principle, we report one novel approach in this communication on getting higher TTA-UC efficiency in self-assembly by controlling the chirality of the acceptors. The acceptors used in this TTA-UC system were designed as the gelators with different chirality, from which three co-gels were obtained. The sensitizer, acceptors, two of the supramolecular assemblies and the circumstances of the two co-gels were all shown in Fig. 1.

|
Download:
|
| Fig. 1. Schematic illustration of the self-assemblies (AR and AR+S) used in the TTA-UC system. PtTPBP was chosen as the low energy photon harvest sensitizer (D, lime ball) and when excited with 635 laser light, triplet state was populated by intersystem crossing (ISC), followed by TTET from donor triplet to accepter triplet in assembly, after the triplet exciton migration and TTA process among the acceptor assembly, upconverted photo luminescence achieved in the self-assembled systems. The racemic assembly with rod like fibers exhibited higher efficiency than homochiral assembly with helical fibers. | |
{kind=link}
We designed a self-assembly system based on 9, 10-bis(phenylethynyl)anthracene (BPEA) derived chiral gelators (AR and AS) as triplet energy acceptors and platinum tetraphenylbenzoporphyrin (PtTPBP) as energy donor. Herein, cyan UC emission could be observed in the self-assembled systems excited by 635 nm. Interestingly, the racemic assemblies composed of R- and S-type acceptor exhibited higher efficiency than homochiral assemblies.
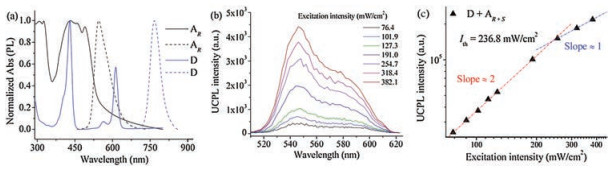
The designed chiral gelators were synthesized by connecting the BPEA moiety with a gelator moiety N, N'-bis(dodecane)- glutamic diamide and the detailed synthetic route was shown in Fig. S1 (Supporting information). Firstly, ultraviolet-visible absorption and fluorescence spectra of the donor and acceptor assemblies were characterized (Fig. 2a). Chiral acceptor AR was chosen as the representative sample. As shown in Fig. 2a, the main peak of fluorescence of the acceptor gel was found at 545 nm, with a shoulder peak around 580 nm, which showed obvious red-shift compared with dilute solution (Fig. S2 in Supporting information). This suggested π-π stacking might be the main driving force for the molecular assembly in the formation process of organogel. After blending with sensitizer (0.029 mmol/L, DMF), the absorption of Q band showed no shift in the co-gel system (Fig. S3 in Supporting information), which suggested that the sensitizer could be molecularly dispersed in the co-gel system. To investigate the TTA-UC process, 635 nm laser was chosen as the excitation power as reported in previous works [34, 35].

|
Download:
|
| Fig. 2. (a) Normalized absorption (solid line) and photo luminescence (dash line) spectra of acceptor AR (black line) and donor D (PtTPBP, blue line); (b) TTA-UC luminescence spectra observed in the co-gels (D + AR+S) under different excitation intensity (λex = 635 nm); (c) The UC intensity of the co-gel (AR+S/D) as a function of the excitation power (635 nm) density. The line was the result of fitting with slope approximate to 2 (red line) and 1 (blue line), Ith was found at 236.8 mW/cm2; The co-gels were all constructed from oxygen-free DMF at room temperature with the same concentration (5.7 mmol/L), the ratio of donor and acceptor was 1:200. | |
{kind=link}
In a standard procedure, the gelator and PtTPBP were mixed in DMF and were heated with oil bath (393.15 K) until it changed to a clear solution. Then, the solution was cooled to room temperature to acquire the stable gel. Cyan light was observed when the gel was irradiated by the red light of 635 nm laser, suggesting the antiStokes shift. UC spectra with different incident power density of 635 nm were investigated. As shown in Fig. 2b, the self-assembled gel formed by racemic acceptors showed UC emission at 545 nm with a shoulder peak around 580 nm, and the emission intensities increased with the increasing of the excitation intensity. The dependence of UC emission intensity on the excitation power density was shown in Fig. 2c. The red and blue lines are the fitting results with slopes of about 2.0 and 1.0 in the low and high excitation intensity ranges, respectively. From the result of the simulation (line fitting), it can be seen that the threshold (Ith) of this TTA-UC system was 236.8 mW/cm2. In addition, the slope was approximate to 2 before the Ith and 1 over the Ith. According to the work of Monguzzi [3], there are two different regimes for the basic physical behavior of TTA process.
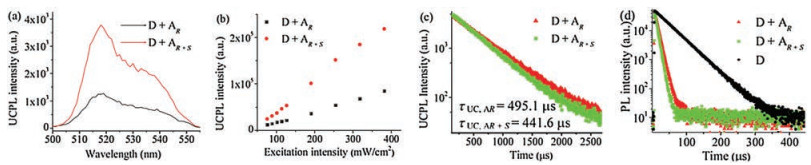
The UC emission was also recorded in the homochiral gels of AR and AS. What is interesting is that the racemic acceptor gel exhibited an obviously TTA-UC emission enhancement compared with the homochiral gels (Figs. 3a and b). At the excitation intensity of 318.4 mW/cm2, the integrated intensity of racemic acceptor gel showed 4-times enhancement. To avoid deviations made by the measurement in the process of spectral acquisition, Fig. 3b showed an average value of three samples for both racemic and homochiral gels. To reveal the reasons for the differences, the detailed parameters accounting for the TTA-UC process were studied.

|
Download:
|
| Fig. 3. (a) TTA-UC spectra of the different co-gel systems (D + AR and D + AR+S) under the same excitation wavelength (635 nm, 318.4 mW/cm2); (b) Integrated TTA-UC emission intensity observed in the co-gels (D + AR and D + AR+S) under different excitation intensity (λex = 635 nm); (c) UC decay (at 545 nm) of the different gels (AR, red; AR + S, green) under 635 nm excitation at room temperature, τUC; AR = 495.1μs, and τUC; AR+S = 441.6μs; (d) Decay of the PtTPBP (0.029 mmol/L) phosphorescence (in oxygen-free DMF solution (black), AR (red) and AR+S(green)). The obtained phosphorescence lifetime was recorded in Table S2.[AR]=[AR+S]=5.7mmol/L, [PtTPBP]=0.029mmol/L. | |
{kind=link}
The lifetime of the fluorescence and phosphorescence of the self-assembled UC systems were collected. The decay curves were shown in Figs. 3c and d, while the simulated and calculated corresponding data were recorded in Tables S1 and S2 (Supporting information). The decay time of upconverted light (τUC) at 545 nm of the co-gels (D + AR and D + AR + S) was characterized under the 635 nm laser excitation. As shown in Fig. 3c, the two lifetimes of the co-gels were τUC; AR = 495.1 μs, and τUC; AR+S = 441.6 μs, respectively. In the process of TTA-UC, τUC related to the fraction of the triplets decay via annihilation (ΦTTA) and the equation [36] as following:

|
where kA = 1/(2 × τUC), IUC is UC emission intensity, TA is the population density of acceptor triplets, kA is the triplet acceptors (AR or AR+S) natural decay rate. The data were fitted with this equation and getting the fractions: ΦTTA; AR = 3.8% and ΦTTA; AR+S = 8.2% separately (Table S1). Besides, the lifetimes of PtTPBP in oxygen-free DMF and in the upconverted gels at 765 nm were collected as shown in Fig. 3d. Obviously, the lifetime of PtTPBP in the co-gel systems was shorter than that in oxygen-free DMF solution.
Accordingly, the transfer efficiency (ΦTTET) can be evaluated from the equation ΦET = 1 - (τ2/τ1) [6]. As Table S2 shown, the calculated efficiencies of the TTET were ΦTTA; AR = 41.2% and ΦTTA; AR+S = 53.1%, respectively. In addition, the racemic gel showed higher TTET efficiency than homochiral gels, which would be the reason of the higher emission intensity observed in the racemic gel. On the other hand, UC quantum yield (UCQY) can be calculated according to equation UCQY = f ×ΦTTET ×ΦTTA ×ΦA/2, in which is the statistical factor accounting for the probability that each TTA event gives rise to a singlet excited state for acceptor and ΦA is the quantum efficiency of the acceptor emission from its singlet states. As the factors of and ΦA were the same in the three UC systems, the equation could be written as: UCQY ∝ ΦTTET ×ΦTTA. For dilute solution, AR, AS, and even the racemic mixture may be showed the same ΦTTA as chirality took no part in TTA process. However, in the assembled systems, different arrangement of chiral molecules would have influence [28, 37, 38]. Therefore, scanning electron microscope (SEM) were introduced to explore the reasons for the enhancement. As shown in Fig. 4a, from the SEM image, the morphology of the co-gels displayed intuitively. The assembled nanostructures of AR were left hand helical fibers while the racemic assembly showed very different morphology. The morphology of the racemic assemblies was rod-like fibers without intense twisting. This phenomenon of morphology was consistent with that described in the previous literatures [39]. In addition, the temperature dependent 1H NMR spectra of the three selfassemblies (AR, AS and AR+S) were collected (Figs. S5–S10 in Supporting information). Fig. S9 illustrated that the binding ability through hydrogen bonding was stronger for racemic assemblies than the homochiral one as the hydrogen on the amide bond for AR +S shifted to lower field at 343.2 K. The stronger hydrogen bonding should be responsible for the better triplet energy transfer as well as stronger UC intensity. We have also measured the circular dichroism spectra of these co-gels. Clearly, homochiral co-gels exhibited mirror-image CD signals while the racemic one showed CD silent (Fig. S11 in Supporting information). The dried xerogel made from the racemic and homochiral co-gels were also investigated (Fig. S12 in Supporting information). Unfortunately, no obvious change could be found, which should be due to the sample fabrication during the drying process of the co-gels. The nanostructures should be broken during the drying process.

|
Download:
|
| Fig. 4. SEM of (a) D + AR assembly (with right hand helical fiber), (b) D + AR+S assembly (rod like fiber). [AR] = [AR+S] = 5.7 mmol/L. | |
{kind=link}
In conclusion, we have realized chiral self-assembly regulated TTA-UC system that is established by chiral acceptor and achiral donor. The formation of racemic co-gel allows efficient UC emission, while the homochiral assemblies result in the poor TTA-UC, in which the racemic assembly showed better molecular packing, which facilitated the triplet energy transfer and migration, enabling the better TTA-UC process. The homochiral system gave rise to the twisted molecular packing, which produce the poor TTET and weak UC. This work which demonstrated that chirality could regulate excited triplet energy transfer will provide deep insight into designing functional UC systems.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (Nos. 21802027, 51673050, 91856115). Thanks for the Youth Foundation of Department of Science and Technology of Jilin Province of China (No. 20160520136JH) and the Scientific Research Project of Education Department of Jilin Province of China (No. 2016319).
Appendix A. Supplementary dataSupplementary material related to this article canbefound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.04.035.
| [1] |
F. Auzel, Chem. Rev. 104 (2004) 139-173. DOI:10.1021/cr020357g |
| [2] |
B. Zhou, B.Y. Shi, D.Y. Jin, X.G. Liu, Nat. Nanotechnol. 10 (2015) 924-936. DOI:10.1038/nnano.2015.251 |
| [3] |
A. Monguzzi, J. Mezyk, F. Scotognella, R. Tubino, F. Meinardi, Phys. Rev. B 78 (2008) 195112. DOI:10.1103/PhysRevB.78.195112 |
| [4] |
Y.Y. Cheng, B. Fueckel, T. Khoury, et al., J. Phys. Chem. Lett. 1 (2010) 1795-1799. DOI:10.1021/jz100566u |
| [5] |
S. Baluschev, V. Yakutkin, T. Miteva, et al., Angew. Chem. Int. Ed. 46 (2007) 7693-7696. DOI:10.1002/anie.200700414 |
| [6] |
A. Monguzzi, M. Frigoli, C. Larpent, R. Tubino, F. Meinardi, Adv. Funct. Mater. 22 (2012) 139-143. DOI:10.1002/adfm.201101709 |
| [7] |
C. Ye, L. Zhou, X. Wang, Z. Liang, Phys. Chem. Chem. Phys. 18 (2016) 10818-10835. DOI:10.1039/C5CP07296D |
| [8] |
J. Wang, Y. Lu, N. McGoldrick, et al., J. Mater. Chem. C 4 (2016) 6131-6139. |
| [9] |
S. Ji, H. Guo, W. Wu, W. Wu, J. Zhao, Angew. Chem. Int. Ed. 50 (2011) 8283-8286. DOI:10.1002/anie.201008134 |
| [10] |
J. Zhao, S. Ji, H. Guo, RSC Adv. 1 (2011) 937-950. DOI:10.1039/c1ra00469g |
| [11] |
C. Reinhard, R. Valiente, H.U. Gudel, J. Phys. Chem. B 106 (2002) 10051-10057. DOI:10.1021/jp0203797 |
| [12] |
M. Haase, H. Schaefer, Angew. Chem. Int. Ed. 50 (2011) 5808-5829. DOI:10.1002/anie.201005159 |
| [13] |
C. Simpson, T.M. Clarke, R.W. MacQueen, et al., Phys. Chem. Chem. Phys. 17 (2015) 24826-24830. DOI:10.1039/C5CP04825G |
| [14] |
S. Baluschev, T. Miteva, V. Yakutkin, et al., Phys. Rev. Lett. 97 (2006) 143903. DOI:10.1103/PhysRevLett.97.143903 |
| [15] |
S.H.C. Askes, A. Bahreman, S. Bonnet, Angew. Chem. Int. Ed. 53 (2014) 1029-1033. DOI:10.1002/anie.201309389 |
| [16] |
V. Gray, D. Dzebo, M. Abrahamsson, B. Albinsson, K. Moth-Poulsen, Phys.Chem. Chem. Phys. 16 (2014) 10345-10352. DOI:10.1039/C4CP00744A |
| [17] |
Q. Liu, T. Yang, W. Feng, F. Li, J. Am. Chem. Soc. 134 (2012) 5390-5397. DOI:10.1021/ja3003638 |
| [18] |
P. Duan, D. Asthana, T. Nakashima, et al., Faraday Discuss. 196 (2017) 305-316. DOI:10.1039/C6FD00170J |
| [19] |
S. Wan, J. Lin, H. Su, J. Dai, W. Lu, Chem. Commun. (Camb.) 54 (2018) 3907-3910. DOI:10.1039/C8CC00780B |
| [20] |
C. Ye, B. Wang, R. Hao, et al., J. Mater. Chem. C 2 (2014) 8507-8514. DOI:10.1039/C4TC00791C |
| [21] |
C. Ye, J. Wang, X. Wang, et al., Phys. Chem. Chem. Phys. 18 (2016) 3430-3437. DOI:10.1039/C5CP05288B |
| [22] |
C.B. Murphy, Y. Zhang, T. Troxler, et al., J. Phys. Chem. B 108 (2004) 1537-1543. DOI:10.1021/jp0301406 |
| [23] |
C. Fan, W. Wu, J.J. Chruma, J. Zhao, C. Yang, J. Am. Chem. Soc. 138 (2016) 15405-15412. DOI:10.1021/jacs.6b07946 |
| [24] |
V. Gray, K. Moth-Poulsen, B. Albinsson, M. Abrahamsson, Coord. Chem. Rev. 362 (2018) 54-71. DOI:10.1016/j.ccr.2018.02.011 |
| [25] |
P.Z. Chen, H. Zhang, L.Y. Niu, et al., Adv. Funct. Mater. 27 (2017) 10. |
| [26] |
H.Q. Peng, L.Y. Niu, Y.Z. Chen, et al., Chem. Rev. 115 (2015) 7502-7542. DOI:10.1021/cr5007057 |
| [27] |
T. Ogawa, N. Yanai, A. Monguzzi, N. Kimizuka, Sci. Rep. 5 (2015) 10882. DOI:10.1038/srep10882 |
| [28] |
D. Yang, P. Duan, M. Liu, Angew. Chem. Int. Ed. 57 (2018) 9357-9361. DOI:10.1002/anie.201804402 |
| [29] |
P.K. Vemula, G. John, Acc. Chem. Res. 41 (2008) 769-782. DOI:10.1021/ar7002682 |
| [30] |
R.V. Ulijn, J. Mater. Chem. 16 (2006) 2217-2225. DOI:10.1039/b601776m |
| [31] |
Y. Murakami, Y. Himuro, T. Ito, et al., J. Phys. Chem. B 120 (2016) 748-755. DOI:10.1021/acs.jpcb.5b09880 |
| [32] |
N. Gan, H. Shi, Z. An, W. Huang, Adv. Funct. Mater. 28 (2018) 1802657. DOI:10.1002/adfm.201802657 |
| [33] |
C.L. Sun, J.F. Xu, Y.Z. Chen, et al., Chin. Chem. Lett. 26 (2015) 843-846. DOI:10.1016/j.cclet.2015.05.030 |
| [34] |
J.H. Kang, E. Reichmanis, Angew. Chem. Int. Ed. 51 (2012) 11841-11844. DOI:10.1002/anie.201205540 |
| [35] |
Q. Liu, B. Yin, T. Yang, et al., J. Am. Chem. Soc. 135 (2013) 5029-5037. DOI:10.1021/ja3104268 |
| [36] |
A. Monguzzi, F. Bianchi, A. Bianchi, et al., Adv. Energy Mater. 3 (2013) 680-686. DOI:10.1002/aenm.201200897 |
| [37] |
A. Sarkar, S. Dhiman, A. Chalishazar, S.J. George, Angew. Chem. Int. Ed. 56 (2017) 13767-13771. DOI:10.1002/anie.201708267 |
| [38] |
R. Sethy, J. Kumar, R. Métivier, et al., Angew. Chem. Int. Ed. 56 (2017) 15053-15057. DOI:10.1002/anie.201707160 |
| [39] |
X. Zhu, Y. Li, P. Duan, M. Liu, Chem Eur. J. 16 (2010) 8034-8040. DOI:10.1002/chem.201000595 |