 2019, Vol. 30
2019, Vol. 30 



Prof. Lijuan Jiao received her Bachelor's degree (2000) from Shandong University, China, and obtained her Master's degree (2003) under the supervision of Prof. Guanwu Wang at University of Science and Technology of China (USTC). She then moved to Louisiana State University, U. S. A., and obtained her Ph.D. (2007) under the supervision of Prof. Kevin M. Smith. She joined Anhui Normal University in 2008, and became a full professor (2010) at College of Chemistry and Materials Science. Her research focuses on the development of novel BODIPY and porphyrin related dyes, understanding their photophysical properties and studying their optoelectrical and biological applications. She received a SPP/JPP Young Investigator Award for her research in BODIPY and oligopyrrole chemistry in 2016.
Conjugated heterocyclic polypyrroles widely exist in natural biomolecules, and play essential roles for the proper function of many biological systems [1-5]. For example, porphyrins (Fig. 1) as the "pigments of life", are the main components of heme and chlorophyll, and are closely associated with the birth, growth and death of many biological systems. Recent research efforts have been devoted to the development of novel porphyrin analogues (porphyrinoids) with advanced novel structures (such as the increased flexibility and cavity), intriguing reactivity (such as the facile inner-ring-annulation and multiple metal-coordination ability), and unique properties (such as tunable aromaticity) that are hard to access from normal porphyrins [1-7].

|
Download:
|
| Fig. 1. The core structures of porphyrins, some representative porphyrinoids and their dimetal complex. | |
{kind=link}
Porphyrins generally contain a tetrapyrrolic core structure, in which the four pyrrole rings are connected to each other via a meso-carbon (the methylene) bridge at the α, α-positions of the pyrrole units. Most reported porphyrinoids are formulated via: 1) the rearrangement of the nitrogen atom in the porphyrin to form confused porphyrins [8-10], such as the N-confused porphyrins (NCP) first independently reported by Furuta [9], Latos-Grażyński and their coworkers [10] (Fig. 1a), 2) the extending the size of the porphyrin to form the so-called "expended porphyrins" [6, 11-13], such as sapphyrin, first discovered by Woodword [11] and then rationally synthesized by Broadhurst and coworkers [12] (Fig. 1b), 3) the replacement of the nitrogen atom(s) with other heteroatoms to form the core-modified porphyrins [14-16], such as the thia-modified porphyrin, first independently reported by Ulman [14], Latos-Grażyński [15] and their coworkers (Fig. 1c), 4) the hybridization of these three strategies to form hybrid porphyrinoids [17-19, 20], for example the thia-modified sapphyrin [17-19] and N-fused pentaphyrin NFP5 first reported by Furuta and coworkers [20] (Fig. 1d). The unique structure provides these novel porphyrinoids unique reactivity and properties and renders them valuable applications as functionalized NIR organic dyes, sensors, fluorescent probes, novel ligands, stable organic radicals, nonlinear optical materials, photosensitizers for photo-dynamic therapy (PDT), and contrasting agents for magnetic resonance imaging (MRI) [1-7, 21-23].
Among those, expanded porphyrinoids have attracted considerable research interests due to their unique multi-metal coordination property (for example, the ability to incorporation of Au and Pt within one macrocycle [24], Fig. 1), and an easily tunable aromaticity. These have been well documented in some recent reviewer articles independently presented by Hiroyuki Furuta, Latos-Grażyński, Osuka, Chandrashekar, D'Souza, Tomé and their coworkers [6, 11, 13, 21-23, 25-30]. Herein, we are mainly focus on some recent synthetic progresses made in multiple-inner-ring fused expanded porphyrins and in the rational synthesis of elusive smaragdyrins.
2. Discovery of N-confused expanded porphyrins and their traditional synthesis from N-confused oligopyrrolesThe first inner-ring-fused expanded porphyrin, N-fused pentaphyrin (NFP5, Scheme 1a) was first isolated by Furuta and coworkers as a side product from the mixture of the classic Rothemund synthesis of porphyrin (an acid catalyzed condensation between pyrrole with aldehyde) [20]. The formation of a fused tripentacyclic inner-ring structure in NFP5 involves the inversion of one pyrrole unit, which undergoes a subsequent nucleophilic attack at the β-position by the nitrogen atom of an adjacent pyrrole in the pentaphyrinogen in situ formed as an intermediate during the DDQ oxidation process.

|
Download:
|
| Scheme 1. N-Confused expanded porphyrins NFP5 (a, b), DNFP5 (b), NFS (c) and oxo-N-confused pentaphyrins ONCP5 and DONCP5 (d) prepared from (a) the classic Rothemund-condensation synthesis of porphyrinoids from pyrrole with aldehyde as a side-product, (b, c) the "confusion strategy" developed by Furuta and coworkers based on an acid-catalyzed condensation of linear oligopyrroles with a middle N-confused pyrrole unit followed by a DDQ oxidation. (d) Confusion strategy failed to generate the corresponding N-fused product, instead was obtained. | |
{kind=link}
These authors rationalized that the replace of one pyrrole unit with an N-confused pyrrole will facilitate this inversion and the subsequent nucleophilic attacking process. Therefore, they for-mulated a one-pot DDQ oxidation of in situ generated N-confused pentaphyrin (from a p-toluenesulfonic acid, p-TSA catalyzed classic [3 + 2] condensation between N-confused tripyrrane and dipyrromethane dicarbinol 2), from which the desired nonaromatic NFP5 was isolated as a major product in 40% yield [31] (Scheme 1b). It is worth noticed that the further DDQ oxidation of NFP5 in dichloromethane at room temperature quantitatively converted it to its aromatic isomer ONFP5, which is unstable and readily fused to form a doubly N-fused porphyrinoids DNFP5 in 80% yield.
Using this confusion strategy, these authors further prepared N-fused-sapphyrin NFS [32] (Scheme 1c) from an oxidative ring fusion of N-confused sapphyrin NCS, generated from a classic [5 + 0] condensation of N-confused pentapyrromethane. It is worth mentioned that although a TFA-catalyzed [3 + 2] conden-sation between 2, 2-bipyrrole and a diol-functionalized tripyrro-methane could also lead to the formation of NCS. In this case, no desired NFS could be generated from a subsequent DDQ oxidation of this resultant NCS (both in the in situ generated form and in the isolated form). Similar to that of N-fused porphyrin (NFP), the further fusion requires an additional bromination step [33].
Although the installation of an N-confused pyrrole unit in the tetra- and pentapyrrolic macrocycles provides a value confusion strategy to form an inner-ring-fusion (a fused tripentacyclic ring) structure in these macrocyclic systems, it is hard to achieve more than three inner ring fusions in the macrocycles. To address this issue, these authors further investigated the possibility of a double confusion strategy to fuse multiple inner rings in the macrocycle system. The installation of two N-confused pyrrole units was smoothly achieved from an acid-catalyzed [3 + 2] condensation between N-confused tripyrromethane and a diol-functionalized N-confused dipyrromethane. However, the subsequent DDQ oxidation failed to generate any fusion products. Instead, only oxo-substituted products were obtained [34] (Scheme 1d).
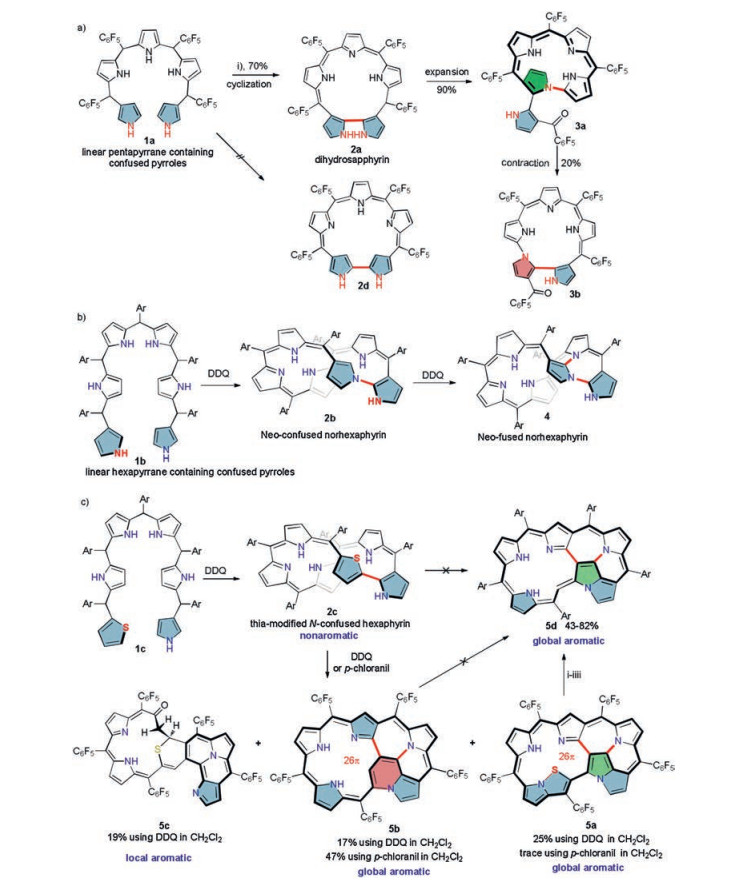
3. Novel strategy to construct N-fused expanded porphyrins with multiple-inner-ring-fusionInspired by the high reactivity of N-confused pyrroles, Xie and coworkers rationalized that the installation of N-confused pyrrole to the terminal of an oligopyrrole would enhance its reactivity and provide an alternative confusion strategy to access inner-ring-fusion. As expected, oligopyrroles 1a and 1b possessing two terminal N-confused pyrrole units indeed show higher reactivity with respect to those previously reported N-confused oligopyrroles. They readily undergo a regioselective oxidative cyclization with DDQ, which leads to the direct formation of pentaphyrin 2a [35] and hexaphyrin 2b [36] without resort to any acid-catalysts (Scheme 2, routes a and b). In addition, the regioselectivity of this cyclization reaction is greatly affected by the length of the starting oligopyrrole. For example, it regioselectively takes place at the two α-positions of the two terminal pyrroles for pentapyrrane 1a to form a β, α-α, β linked bipyrrole unit in dihydrosapphyrin 2a. By contrast, the two terminal pyrroles are connected to each other at the N, α-positions for hexapyrrane 1b to form a β, N-α, β bipyrrole fragment in neo-confused norhexaphyrin 2b.

|
Download:
|
| Scheme 2. Oxidative cyclization of linear oligopyrroles 1 containing terminal N-confused pyrrole(s) to form novel expanded porphyrinoids 2 with different linked bipyrrole unit (β, α-α, β, α, α-N, β, N, α-α, β, β, N-α, β and β, α-N, α) and their further macrocycle transformation reaction (including multiple-inner-ring fusion) reported by Xie and coworkers [35-37]. Reaction conditions: (a) O2 in Et3N/MeCN (5d in 43% yield), (b) H2O2/AcOH in THF (5d in 55% yield), (c) mCPBA in CH2Cl2, 5d in 62% yield, (d) PPh3 in CH2Cl2 (5d in 82% yield). | |
{kind=link}
In addition, the N-confused pyrrole in these resultant expanded porphyrinoids is also highly reactive and readily undergoes oxidative C-H elimination. Among those, it promotes a ring expansion/contraction reaction for dihydrosapphyrin 2a to form pyrrolyl norrole 3a and isosmaragdyrin analogue 3b containing a unique α, α-N, β and a β, α-N, α bipyrrole, respectively. For neo-confused norhexaphyrin 2b, it initiates an interesting inner-ring fusion reaction to generate an unprecedent multiple inner-ring fusion. Unlike the formation a 5, 5, 5-tricyclic structure in N-fused porphyrinoids previously reported by Furuta and coworkers [20, 31-33], this inner-ring-fusion brings the formation of an unprecedented 5, 5, 5, 7-tetracyclic structure in 4.
Considering the high resistance of thiophene moiety toward oxidative C-H elimination and the relatively easy breakage/cleavage of C-S bond not offered by pyrrole, this group has lately incorporated a terminal thiophene moiety to the starting linear oligopyrrole 1c [37] (Scheme 2c). Upon oxidative ring cyclization, a thia-modified N-confused hexaphyrin 2c is obtained. The simple replacement of one terminal N-confused pyrrole with thiophene moiety indeed brings different regioselectivity in this oxidative cyclization and the subsequent oxidative inner-ring-fusion reactions. The cyclization regioselectively occurs at the α, α-positions of the two terminal units to form a α, α-α, β-linked biheterocycle unit. The subsequent inner-ring-fusion takes place (using either DDQ or p-chloranil) via an oxidative skeletal rearrangement accompanied with the thiophene cleavage and desulfurization processes, from which the globally aromatic thiopyrrolopentaphyrin 5a and a sulfur-free indolizine-containing pentaphyrin 5b are generated.
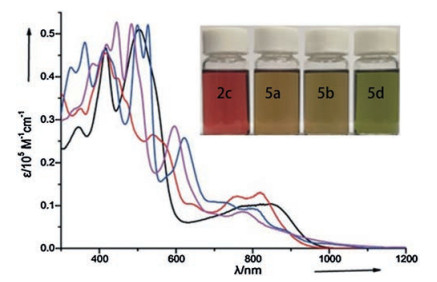
The usage of DDQ also leads to the formation of a locally aromatic product 5c, which is not observed by using p-chloranil. The attempt to further oxidize 5b with DDQ failed at varying conditions. On the other hand, the desulfurization of 5b proceeded by treating it in CH3CN under air in the presence of Et3N. The usage of mCPBA as the oxidant further improved yield of 5d to 62%. The highest yield (82%) for 5d is obtained under a reductive elimination condition (using PPh3 in CH2Cl2). Both attempts to generate 5d from the direct oxidation of 2b or 5b failed. These resultant inner-ring-fusion products 5 all show broad NIR bands (up to 1000 nm, Fig. 2) and weak NIR absorption emissions (Φ≤ 0.18%) similar to that of the non-annulated 2c. The synthetic strategy reported by this group provides a facile access to a variety of expanded porphyrinoids, not only to those containing different bipyrrole linkage (β, α-α, β, α, α-N, β, N, α-α, β, β, N-α, β and β, α-N, α), but also to those containing unprecedented poly-inner-ring annulations.

|
Download:
|
| Fig. 2. Overlaid absorption spectra of 2c (black line), 5a (red line), 5b (blue line) and 5d (pink line) in CH2Cl2. Inset: pictures of the CH2Cl2 solutions of these compounds at ambient light. Reproduced with permission [37]. Copyright 2019, Wiley Publishing Group. | |
{kind=link}
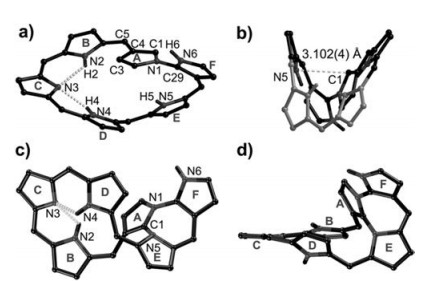
These multiple-inner-ring-fusion products show interesting conformational behavior varying with the size of the macrocycle and the substitution. Among those, Norhexaphyrin 2b shows a figure-of-eight overall structure with a dihedral angle of 52.08° with the N-confused pyrrole ring (Fig. 3) [36]. Its fusion product 4 also exhibits a folded conformation with a boat conformation (with the A/E, E/F, and F/A interplane angles of 61.58°, 35.58° and 39.68°, respectively) for the newly formed seven-membered ring. The thiophene-modified 2c adopts a similarly figure-of-eight overall structure with the thiophene ring located at the hinge of this figure-of-eight structure (a dihedral angle of 41.48° with the N-confused pyrrole ring) (Fig. 4) [37]. The highly distorted structures of 2b and 2c indicate the nonaromaticity nature of these two compounds. The formation of multiply C-C/N fused coplanar structures in 5a, 5b and 5c was clearly observed in their X-ray structures. Among those, a moderate derivation (dihedral angle, 47.28°) of the new formed pyrrolo[1, 2]isothiazole unit (rings G/E) from the mean plane composed of all the remaining units of the macrocycle was observed in 5a, which may be attributed to the steric hindrance effect from a large sized sulfur atom within the core. By contrast, the fused pentaphyrin 5b shows a more planar structure than that of 5a. This may associate with the cleavage of bulky sulfur atom. In addition, the formation of a locally fused 14π-conjugated structure with an annulated 2H-thiopyran moiety and a keto-bridged sub-cyclic unit in 5c are also visible in its X-ray structure.

|
Download:
|
| Fig. 3. Complementary views of the molecular structures of 2b (a, b) and 4 (c, d). C6F5 and hydrogen atoms attached to carbons are omitted for clarity. Reproduced with permission [36]. Copyright 2014, Wiley Publishing Group. | |
{kind=link}

|
Download:
|
| Fig. 4. Complementary views of the molecular structures of 2c (a, b), 5a (c, d), 5b (e, f) and 5c (g, h). C6F5 and hydrogen atoms attached to carbons are omitted for clarity. Reproduced with permission [37]. Copyright 2019, Wiley Publishing Group. | |
{kind=link}
4. Novel strategy to construct smaragdyrins by SNAr reaction
Another hardly accessible expanded porphyrinoid is smaragdyrins (Scheme 3), which belongs to a family of pentapyrrolic macrocyclic system. Smaragdyrin is first isolated as a byproduct from the synthesis of sapphyrin. Recently, many synthetic progresses have been made to the facile synthesis of many pentapyrrolic macrocycles [6, 11, 13, 21-23, 25-30]. For example, [22]sapphyrins have been well known as fluoride anion sensors extensively studied by Sessler and coworkers [38-40], and the well-established synthesis of β-decaalkyl-substituted [22]penta-phyrin(1.1.1.1.1)s can be dated back to 1980s [41] (Fig. 1). Even some of its analogue or isomers, like the core-modified smaragdyrins and [22]isosmaragdyrin have been well studied. For example, the rational synthesis of the core-modified smaragdyrins has recently been accomplished by Chandrashekar and has been identified as a planar aromatic molecule for anion sensing [17, 42-44], and [22] isosmaragdyrin has been prepared in its HCl salt form by Sessler and coworkers (Fig. 1) [45]. By contrast, the rational synthesis of smaragdyrins remains a mystery before a very lately report by Song and coworkers.

|
Download:
|
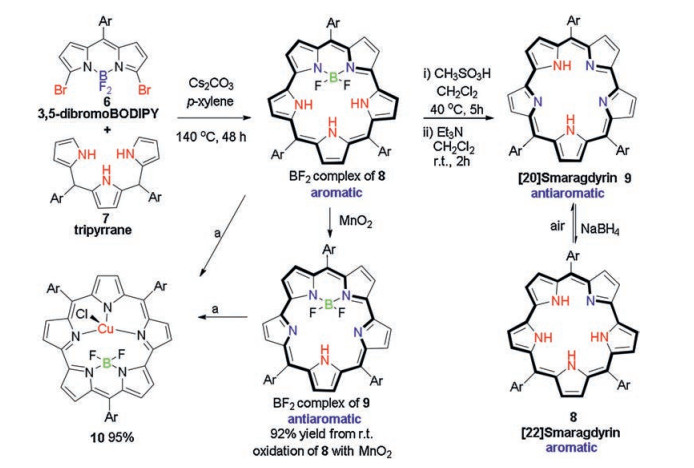
| Scheme 3. The synthesis of [22]smaragdyrin 8 and [20]smaragdyrin 9 based on a straightforward double SNAr reaction on readily available 3, 5-dibromoBODIPY 6, the facile tune of the aromaticity of the system via simple BF2 complexation/decomplexation, and the further metal coordination for the BF2 complexes of these smaragdyrins disclosed by Song and coworkers [50]. Reaction condition a: CuCl2/NaOAc in CH2Cl2 at room temperature. | |
{kind=link}
Inspired by a lately reported new synthetic strategy to open-chain linear oligopyrroles based on a straightforward SNAr reaction on readily accessible 3, 5-dibromoBOIPYs with pyrroles [46-49], Song and coworkers have lately prepared [22]smaragdyrin 8 and its reduced analogue [20]smaragdyrin 9 [50] (Scheme 3). The double SNAr reaction of 3, 5-dibromoBOIPY 6 with tripyrrane 7 smoothly generated the desired [22]smaragdyrin 8 as a BF2 complex in a high yield. Interestingly, the BF2 cleavage of this complex with methanesulfonic acid under nitrogen atmosphere and a subsequent work-up with triethylamine did not generate the expected free base form of [22]smaragdyrin 8. Instead, the oxidized analogue [20]smaragdyrin 9 was mainly generated. Although a further reduction of this resultant 9 with NaBH4 can smoothly generate 8 in nearly quantitative yield, this resultant [22]smaragdyrin 8 is unstable in its free base form in the air and readily converted back to the starting 9 on a silica gel purification process. This instability has been attributed to the Coulombic repulsion among the four pyrrolic NH protons.
The BF2 complex of [20]smaragdyrin 8 is achieved in a high yield from the BF2 complex of its analogue, 9 via an oxidation using MnO2. In addition, [22]smaragdyrin 9 can also be easily oxidized by CuCl2 in the presence of NaOAc in CH2Cl2 since the BF2 complex of 9 readily coordinated with Cu(Ⅱ) to form a Cu(Ⅱ)-BF2 complex of [20]smaragdyrin (10) in a high yield at room temperature, similar to that of its BF2 analogue of 8 (Scheme 3).
Interestingly, this BF2 complexation/decomplexation has been found to be a facile way to tune the stability of these smaragdyrins. In the free base form, [20]smaragdyrin 9 is a stable antiaromatic compound, while its reduced form, [22]smaragdyrin 8 is an unstable aromatic one. Upon BF2 complexation, a reversible stability of these two compounds was observed. This interesting, facile tune of the aromaticity via variation of the oxidation state of the macrocycle is in good agreement with those previously reported expanded porphyrinoids [6]. The stability reversal by simply BF2 complexation/ decomplexation is unique among those reported pentaphyrins related systems.
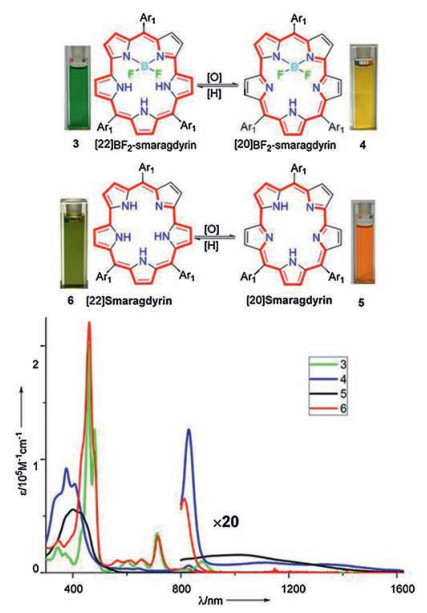
The different aromaicity between the 8 (and its BF2 complex), and 9 (and its BF2 complex) is in good agreement with their absorption and their dramatically different solution colors at ambient (Fig. 5). Similar to those of previously reported aromatic core modified smaragdyrins [42-44], [22]smaragdyrin 8 shows a sharp Soret-band at 460 nm and weak Q-bands at 566, 613, 653, 715 and tailed to 812 nm. Its BF2 complex shows a similar sharp but splitted Soret-band at 455 and 478 nm, and red-shifted weak Q-bands at 600, 650, 710 and 879 nm. Upon excitation at 480 nm, this BF2 complex shows characteristic fluorescence emission at 727 and 800 nm. By contrast, [20]smaragdyrin 9 and its complex show dramatically different absorption spectra from those of 8 and its complex. A broad and ill-defined absorption bands are observed in [20]smaragdyrin 9 and its complex within the range of 376 nm to 600 nm with long tails in the NIR regions. This indicates the antiaromaticity of these compounds. In addition, no appreciable fluorescence peaks were observed for [20]smaragdyrin 9 and its complex due to their optically forbidden lowest transition state.

|
Download:
|
| Fig. 5. The solution pictures of 8 ([22]smaragdyrin), 9 ([20]smaragdyrin) and their BF2 complexes in chloroform under ambient light condition (top) and their overlaid absorption spectra (bottom): 8 (red), 9 (black) and the BF2 complexes (green for 8 and blue for 9) in CHCl3. Reproduced with permission [50]. Copyright 2018, American Chemical Society. | |
{kind=link}
5. Conclusion
In summary, novel expanded porphyrinoids containing innerring-fusion or tunable aromaticity show intriguing properties and have attracted wide research interests in many research fields. Considering research efforts have been devoted to the efficient construction of these molecules. Among those, Xie, Song and their coworkers have independently reported a set of novel expanded porphyrinoids with unique multiple-inner-ring-fusion and smaragdyrins with facile tunable aromaticity, respectively. The synthesis is based on an oxidative ring-cyclization and a subsequent ring-fusion reaction on N-confused pyrrole containing linear oligopyrroles or a straightforward double SNAr reaction on readily available 3, 5-dibromoBODIPY. These new synthetic strategies use mild reaction conditions and have good compatibility with various functionalities. It provides great tools to access many higher homologues in expanded porphyrinoids with advanced multiple inner-ring-fusion structure. Further research in this area may involve the investigation of the application of these new synthetic/fusion strategies in preparing multiple inner-ring fused expanded porphyrins, like multiple-inner-ring fused heptaphyrins and octaphyrins, and their metal coordination properties. In addition, considering the strong long-wavelength NIR absorption property of these novel porphyrinoids (up to NIR Ⅱ region) and the relatively low fluorescence emission properties, the investigation of their potential therapeutical applications, for example as novel NIR photosensitizers and photothermal agents for the photodynamic and photothermal therapy of tumors could also be the future research direction in this area.
AcknowledgmentWe thank the National Nature Science Foundation of China (Nos. 21672006, 21672007 and 21871006) for supporting this work.
| [1] |
(a) C. Li, J. Zhang, J. Song, et al., Sci. China Chem. 61 (2018) 511-514; (b) B. Yin, X. Liang, W. Zhu, et al., Chin. Chem. Lett. 29 (2018) 99-101. |
| [2] |
(a) Y. Ning, J. Tang, Y.W. Liu, et al., Chem. Sci. 9 (2018) 3742-3753; (b) W. Miao, E. Dai, W. Sheng, et al., Org. Lett. 19 (2017) 6244-6247. |
| [3] |
(a) X. Yang, P. Zhu, J. Ren, et al., Chem. Commun. 55 (2019) 1201-1204; (b) W. Sheng, F. Lv, B. Tang, et al., Chin. Chem. Lett. 30 (2019) 1825-1833. |
| [4] |
K. Wang, D. Qi, Y. Li, et al., Coord. Chem. Rev. 378 (2019) 188-206. DOI:10.1016/j.ccr.2017.08.023 |
| [5] |
F. Wu, J. Liu, P. Mishra, et al., Nat. Commun. 6 (2015) 7547-7555. DOI:10.1038/ncomms8547 |
| [6] |
B. Szyszko, M.J. Białek, E. Pacholska-Dudziak, L. Latos-Grażyński, Chem. Rev. 117 (2017) 2839-2909. DOI:10.1021/acs.chemrev.6b00423 |
| [7] |
Y. Ding, W.H. Zhu, Y. Xie, Chem. Rev. 117 (2017) 2203-2256. DOI:10.1021/acs.chemrev.6b00021 |
| [8] |
M. Toganoh, H. Furuta, Chem. Lett. 48 (2019) 615-622. DOI:10.1246/cl.190250 |
| [9] |
H. Furuta, T. Asano, T. Ogawa, J. Am. Chem. Soc. 116 (1994) 767-768. DOI:10.1021/ja00081a047 |
| [10] |
P.J. Chmielewski, L. Latos-Grażyński, K. Rachlewicz, T. Glowiak, Angew. Chem. Int. Ed. 33 (1994) 779-781. DOI:10.1002/anie.199407791 |
| [11] |
A. Srinivasan, H. Furata, Acc. Chem. Res. 38 (2005) 10-20. DOI:10.1021/ar0302686 |
| [12] |
M.J. Broadhurst, R. Grigg, A.W. Johnson, J. Chem. Soc. Perkin Trans. 1 (1972) 2111-2116. DOI:10.1039/P29720001124 |
| [13] |
F. D'Souza, Angew. Chem. Int. Ed. 54 (2015) 4713-4714. DOI:10.1002/anie.201501046 |
| [14] |
A. Ulman, J. Manassen, J. Am. Chem. Soc. 97 (1975) 6540-6544. DOI:10.1021/ja00855a042 |
| [15] |
L. Latos-Grażyński, Core-modi fied heteroanalogues of porphyrins and metalloporphyrins, in: K.M. Kadish, K.M. Smith, R. Guilard (Eds.), The Porphyrin Handbook, Academic Press, San Diego, 2000.
|
| [16] |
A. Singhal, Top. Curr. Chem. 376 (2018) 21-39. DOI:10.1007/s41061-018-0199-y |
| [17] |
J.L. Sessler, J.M. Davis, Acc. Chem. Res. 34 (2001) 989-997. DOI:10.1021/ar980117g |
| [18] |
N. Sprutta, L. Latos-Grażyński, Org. Lett. 3 (2001) 1933-1936. DOI:10.1021/ol0159773 |
| [19] |
D. Wu, A.B. Descalzo, F. Weik, et al., Angew. Chem. Int. Ed. 47 (2007) 193-197. DOI:10.1002/anie.200702854 |
| [20] |
J.Y. Shin, H. Furuta, A. Osuka, Angew. Chem. Int. Ed. 40 (2001) 619-621. DOI:10.1002/1521-3773(20010202)40:3<619::AID-ANIE619>3.0.CO;2-X |
| [21] |
M. Stepień, N. Sprutta, L. Latos-Grażyński, Angew. Chem. Int. Ed. 50 (2011) 4288-4340. DOI:10.1002/anie.201003353 |
| [22] |
B. Szyszko, L. Latos-Grażyński, Chem. Soc. Rev. 44 (2015) 3588-3616. DOI:10.1039/C4CS00398E |
| [23] |
L. Costa, J. Costa, A. Tomé, Molecules 21 (2016) 320. DOI:10.3390/molecules21030320 |
| [24] |
S. Gokulnath, K. Yamaguchi, K. Toganoh, et al., Angew. Chem. Int. Ed. 50 (2011) 2302-2306. DOI:10.1002/anie.201006784 |
| [25] |
S. Saito, A. Osuka, Angew. Chem. Int. Ed. 50 (2011) 4342-4373. DOI:10.1002/anie.201003909 |
| [26] |
H. Furuta, H. Maeda, A. Osuka, Chem. Commun. 17 (2002) 1795-1804. DOI:10.1039/B200525P |
| [27] |
M. Toganoh, H. Furuta, Chem. Commun. 48 (2012) 937-954. DOI:10.1039/C1CC14633E |
| [28] |
J. Shin, K.S. Kim, M.C. Yoon, et al., Chem. Soc. Rev. 39 (2010) 2751-2767. DOI:10.1039/b925417j |
| [29] |
R. Misra, T.K. Chandrashekar, Acc. Chem. Res. 41 (2008) 265-279. DOI:10.1021/ar700091k |
| [30] |
T.K. Chandrashekar, S. Venkatraman, Acc. Chem. Res. 36 (2003) 676-691. DOI:10.1021/ar020284n |
| [31] |
A. Srinivasan, T. Ishizuka, H. Furuta, Angew. Chem. Int. Ed. 43 (2004) 876-879. DOI:10.1002/anie.200352946 |
| [32] |
I. Gupta, A. Srinivasan, T. Morimoto, M. Toganoh, H. Furuta, Angew. Chem. Int. Ed. 47 (2008) 4563-4567. DOI:10.1002/anie.200705984 |
| [33] |
H. Furuta, T. Ishizuka, A. Osuka, T. Ogawa, J. Am. Chem. Soc. 121 (1999) 2945-2946. DOI:10.1021/ja9902672 |
| [34] |
A. Srinivasan, T. Ishizuka, H. Maeda, H. Furuta, Angew. Chem. Int. Ed. 43 (2004) 2951-2955. DOI:10.1002/anie.200453732 |
| [35] |
Y. Xie, P. Wei, X. Li, et al., J. Am. Chem. Soc. 135 (2013) 19119-19122. DOI:10.1021/ja4112644 |
| [36] |
P.C. Wei, K. Zhang, X. Li, et al., Angew. Chem. Int. Ed. 53 (2014) 14069-14073. DOI:10.1002/anie.201408307 |
| [37] |
Q. Li, M. Ishida, H. Kai, et al., Angew. Chem. Int. Ed. 58 (2019) 5925-5929. DOI:10.1002/anie.201900010 |
| [38] |
J.L. Sessler, M.J. Cyr, V. Lynch, E. McGhee, J.A. Ibers, J. Am. Chem. Soc. 112 (1990) 2810-2813. DOI:10.1021/ja00163a059 |
| [39] |
P.J. Chmielewski, L. Latos-Grażyński, K. Rachlewwicz, Chem. -Eur. J. 1 (1995) 68-73. DOI:10.1002/chem.19950010111 |
| [40] |
H. Rexhausen, A. Gossauer, J. Chem. Soc. Chem. Commun. 6 (1983) 275-275. DOI:10.1039/c39830000275 |
| [41] |
T. Chatterjee, A. Srinivasan, M. Ravikanth, T.K. Chandrashekar, Chem. Rev. 117 (2017) 3329-3376. DOI:10.1021/acs.chemrev.6b00507 |
| [42] |
Y. Pareek, M. Ravikanth, T.K. Chandrashekar, Acc. Chem. Res. 45 (2012) 1801-1816. DOI:10.1021/ar300136s |
| [43] |
S.J. Narayanan, B. Sridevi, T.K. Chandrashekar, U. Englich, K. Ruhland-Senge, Org. Lett. 1 (1999) 587-590. DOI:10.1021/ol990108n |
| [44] |
S.J. Narayanan, B. Sridevi, T.K. Chandrashekar, A. Vij, R. Roy, J. Am. Chem. Soc. 121 (1999) 9053-9068. DOI:10.1021/ja991472k |
| [45] |
J.L. Sessler, J.M. Davis, V. Lynch, J. Org. Chem. 63 (1998) 7062-7065. DOI:10.1021/jo981019b |
| [46] |
T. Jiang, P. Zhang, C. Yu, et al., Org. Lett. 16 (2014) 1952-1955. DOI:10.1021/ol500507f |
| [47] |
C. Yu, L. Jiao, X. Tan, et al., Angew. Chem. Int. Ed. 51 (2012) 7688-7691. DOI:10.1002/anie.201202850 |
| [48] |
E. Dai, W. Pang, X. Zhang, et al., Chem. -Asian J. 10 (2015) 1327-1334. DOI:10.1002/asia.201500118 |
| [49] |
J. Li, Q. Zhang, J. Yin, et al., Org. Lett. 18 (2016) 5696-5699. DOI:10.1021/acs.orglett.6b02924 |
| [50] |
D. Xie, Y. Liu, Y. Rao, et al., J. Am. Chem. Soc. 140 (2018) 16553-16559. DOI:10.1021/jacs.8b07973 |