 2019, Vol. 30
2019, Vol. 30 


Most organic luminescent materials contain double-bond molecules, such as aromatic compounds and polyene compounds. The delocalized π in these materials is most closely related to luminescence. Each π bond contains two electrons with opposite spins. Therefore, the ground state of the π bond system is a singlet state, and the excited state can be a singlet state or a triplet state. According to Hund's rule, the singlet-state energy level is always higher than the corresponding triplet-state energy level. The transition between the singlet state and the triplet state is forbidden; hence, the absorption (excitation) process occurs mostly during the transition from the ground state S0 to the singlet state S1, S2, , and the electrons in the excited state can return to the ground state S0 through several ways. The electrons in the high excited state rapidly relax to the lowest excited state S1, and then luminescent transition of S1→S0 occurs, which is called fluorescence; whereas the electrons in the singlet state are transferred to the triplet state via the intersystem crossing relaxation without radiation. The luminescent transition from T1 back to the ground state S0 is called phosphorescence. This is the basic work (luminescence) process of photoluminescent (PL) compounds [1, 2]. The luminescence properties of organic luminescent materials are closely related to molecular arrangement, flexibility of conformation change, and interaction between molecules. Thus, many stimulations that can lead to molecular stacking and conformation changes will affect the energy levels of HOMO and LUMO [3].
Pressure is one of the most important thermodynamic parameters that affect the structure and properties of materials [4-13]. High pressure can effectively increase the degree of molecular accumulation, change the interaction between molecules, affect the arrangement and overlap of the electron clouds between molecules or between intramolecular electron clouds, improve the energy system, reduce the stability of the system, and affect the molecular conformation, making structural mutation and chemical reaction of the system possible [14]. High pressure plays a vital role in the study on organic luminescent materials, which are materials of considerable interest in discovering new luminescence phenomena and exploring the mechanism of luminescence. Given the technological advances in the studied on high pressure over the past few years, especially the diamond anvil cell (DAC) method, high-pressure luminescence has been widely studied by scientists and technicians [15].
DAC is an ideal tool for studying the behavior of luminescent materials under high pressure because diamond has a low birefringence and good spectral permeability. DAC uses many kinds of structural types, but the structures are comprise three parts: diamond anvil, gasket, and support guide device [16]. Samples are placed in the metal gasket hole between the flat and parallel culets of two diamond anvils. When the two opposed anvils are pushed together by external force, the sample is subjected to high pressure. Hydrostatic pressure conditions can be obtained by filling the pressure chamber with liquid-pressuretransmitting medium [17]. The pressure in the DAC sample room is calibrated by the fluorescence spectra of rubies containing approximately 0.5% chromium [18]. Many kinds of in situ characterizations based on DAC, including Raman scattering, infrared, X-ray diffraction (XRD), ultraviolet–visible (UV–vis) absorption, and fluorescence spectroscopy, gain significant nsights into the pressure-induced changes in intermolecular interactions. These high-pressure techniques provide opportunities for examining organic luminescent material systems with unprecedented precision, accuracy, and sensitivity [19].
Over the past few years, a series of interesting high-pressure fluorescence phenomena has been discovered and successfully studied by applying high pressure. Researchers are particularly concerned with pressure-responsive luminescent materials, and the study of such materials explores the mechanism of matter luminescence and its potential applications [20-25]. During the study, some materials demonstrated pressure-induced emission (PIE) or pressure-induced enhanced emission (PIEE) phenomena. Currently, PIE materials are found in halogen perovskite materials [26-31] and semiconductor nanocrystals [32, 33]. PIEE materials are also found in organic luminescence materials [34-43].
Nowadays, given that piezochromic luminescent materials and pressure-induced emission materials have many potential applications in force sensors, anticounterfeiting, data recording and storage, light-emitting devices, probes, and so on, the study of pressure-responsive luminescence and PIEE remains the concern of many researchers [37, 44, 45]. The present work highlights the recent advances in piezochromic luminescence and pressureinduced emission by using in situ high-pressure techniques and briefly reviews the new development themes of related areas.
2. Piezochromic luminescent materialsThe color of molecular luminescence can be changed by force in two ways: One is by changing the chemical structure of the molecule involving the fracture of the old bond and the formation of a new bond, which essentially forms a new molecule and emits a different light; the other way is by changing the physical aggregation state. Under the action of external forces, the molecule stacking mode, molecular conformation, or intermolecular interaction change, affecting the energy level of the molecule and leading to the change in luminescence [46-48]. Isotropic compression has a unique effect on crystals, because extremely highly isotropic hydrostatic pressure can significantly change the molecular conformation and aggregation state of crystals without destroying the crystal shape [49, 50].
2.1. Anthracene derivativesAnthracene is a common polycyclic aromatic hydrocarbon with a wideband gap. Its various derivatives have been widely studied in the field of organic luminescence. Hence, the derivatives of anthracene must be carefully selected in the study of the effects of high pressure on organic luminescent materials. In 2012, Tian et al. [51] studied 9, 10-bis((E)-2-(pyrid-2-yl)vinyl)anthracene (BP2VA, shown in Fig. 1a). By applying pressure through DAC, the BP2VA powder emission exhibited a significant red shift from 528 nm to 652 nm (Fig. 1b). To study the relationship between the molecular aggregation state (induced by high pressure) and the luminescent properties, Tian et al. obtained three crystals (C1, C2 and C3, shown in Figs. 1c–e), and their respective luminescence properties are shown in Fig. 1f. The red shift of the BP2VA powder under high pressure is found to be related to the three crystals. Through structural analysis, C1 and C2 were found to adopt J-type aggregation along the y-axis and H-type aggregation along the x-axis, respectively; whereas C3 was found to form a dimer in a tight face-to-face aggregation along the x-axis. The overlap of anthracene planes between the adjacent molecules increased from C1 to C3. The anthracene core is responsible for intramolecular radiative HOMO→LUMO transition, so the stacking mode and intermolecular interaction of the anthracene plane affect the HOMO→LUMO band gap and the luminescence of the crystalline. According to comprehensive analysis, the anthracene planes of two adjacent molecules exhibit nearly no overlap in C1 without π–π interaction. In C2, the anthracene planes of the adjacent molecules overlap by approximately 40%, forming a weak π–π interaction. As for C3, adjacent anthracene planes are almost stacked face-to-face with strong π–π interaction. Finally, as the π–π interaction strengthens, the BP2VA redshift emission under pressure occurs. The synchrotron XRD of the sample was analyzed, as shown in Fig. 1g. The refined result shows that the a-and c-axes of the BP2VA molecular crystal exhibit normal compression, whereas the b-axis exhibits negative compression. This finding fully demonstrates that the b-axis of the molecule is elongated under high pressure. Thus, the physicochemical mechanism of discoloration involves the stretching of the aggregate behavior from the J aggregates along the b-axis to the H aggregates and then to the dimer, which only existed at high pressure. Such a change leads to a wide range of redshift emission.

|
Download:
|
| Fig. 1. (a) Molecular structure of BP2VA. (b) PL spectrum of BP2VA powder under external pressure (FI = fluorescence intensity).Stacking modes of the anthracene planes in adjacent BP2VA molecules in three single crystals: (c) C1, (d) C2, and (e) C3. (f) PL spectra of the three crystals. (g) Change of lattice constants with pressure. Reproduced with permission [51]. Copyright 2012, Wiley. | |
{kind=link}
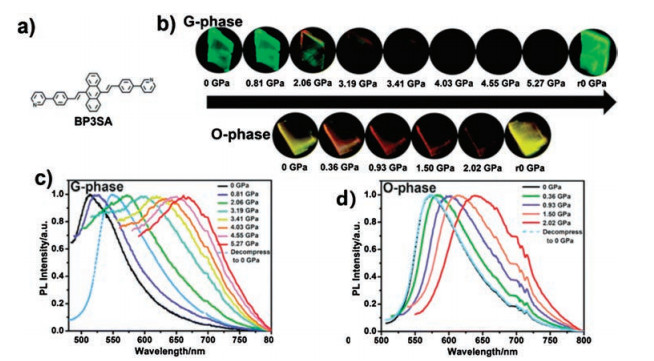
Similarly, Tian et al. [52] studied 9, 10-bis((E)-2-(pyridin-4-yl) vinyl)anthracene (BP4VA), and the BP4VA powder emission red shifted under high pressure (from 523 nm to 650 nm). From the two crystalline analyses conducted, the main interaction between adjacent molecules in C1 (emission at approximately 510 nm) is CH- π interaction and adopts J-type aggregation along the X state. Given that the central anthracene planes do not overlap, no π–π interaction is formed in C1. C2 (emission at approximately 550 nm) exhibits an H-type aggregation along the y-axis. A great overlap occurs in the central anthracene plane; hence, a strong π–π interaction exists in C2. Therefore, BP4VA piezochromic behavior is still attributed to the change in the molecular aggregation state under high pressure. Based on previous studies, Tian et al. [53] further investigated the piezochromic behavior of 9, 10-bis-((E)-4- (pyridin-3-yl)styryl)anthracene (BP3SA) under different pressures. Two types of crystals were obtained: G-phase (triclinic) and Ophase (monoclinic). Both G-phase (from 511 nm to 660 nm) and Ophase (from 575 nm to 641 nm) exhibit significant red shift under high pressure (Fig. 2), but when the pressure is released, the following changes occur: G-phase emission is partially restored, and O-phase is completely restored to its original state. These phenomena can be explained by the interaction of crystalline molecules. For G-phase, various high-intensity C–H⋯N interactions can help strengthen the molecular configuration and lock molecular rotation, which can resist the effects of the external pressure. Thus, G-phase shows low sensitivity to pressure. On the contrary, O-phase can respond sensitively to pressure due to the lack of C–H⋯N interaction.
Moreover, given that the dimer was formed by aggregation, Yang et al. [54] found that 2-(anthracen-9-yl)thianthrene (2-TAAN) exhibits a different behavior from common anthracene derivatives, which exhibit piezochromic behavior. From 0.12 GPa to 1.70 GPa, only 2-TA-ANshowsa decrease in emission. Then a continuous red shift occurs at 1.70 GPa to 9.41 GPa. The change in this case is due to the π–π interaction caused by the overlap of the anthracene planes described above. The synchrotron XRD and theoretical calculations explain this phenomenon. From 1 atm to 1.70 GPa, the excited dimer always relaxes itself to the same excimer equilibrium geometry before emission (no redshift). From 1.70 GPa to 9.41 GPa, the excimer no longer relaxes itself to its equilibrium geometry before emission (redshift) due to the restriction of external pressure. Most importantly, this turning point of fluorescence redshift at 1.70 GPa indicates the π–π interplanar distance of excimer equilibrium geometry.

|
Download:
|
| Fig. 2. (a) Structure of the BP3SA. (b) Images of BP3SA G-phase and O-phase under different applied pressures ("r0" represents "release pressure to 0"). (c, d) PL spectra of G phase and O-phase under different applied pressures. The excitation wavelength is 365 nm. Reproduced with permission [53]. Copyright 2019, The Royal Society of Chemistry. | |
{kind=link}
In summary, the piezochromic behavior of anthracene derivatives originates from the changes in their aggregation state, indicating that the latter is one of the important ways to regulate the change in luminescence. Thousands of anthracene derivatives and new phenomena are yet to be discovered in the field of piezochromic materials.
2.2. Triphenylamine (TPA) derivativesTPA is a typical nonplanar molecule that is important for studies on organic light-emitting materials. It has a large conjugate plane, and the single molecule has a tetrahedral configuration. Three benzene rings are linked to the central nitrogen atom via a C–N single bond, allowing the benzene ring to rotate freely. TPA and its derivatives have a wide range of applications in the field of organic luminescence. Ma et al. [22] found that D-A-substituted TPA derivative has high fluorescence efficiency in the solid state, largely due to the hybridized local and charge-transfer state (HLCT). They conducted a study on (Z)-4-(2-cyano-2-(4'-(diphenylamino)-[1, 1'-biphenyl]-4-yl)vinyl)benzonitrile (pCN-TPA), which consists of twisted diphenylacrylonitrile (DiCN) as acceptor moiety and bulky TPA group as donor moiety. pCN-TPA has distinct intramolecular charge-transfer (ICT) properties and has a high fluorescence efficiency in the aggregated state. In this kind of material, the locally excited (LE) HLCT state provides a high radiative transition rate for high PL efficiency quantum yields (PLQYs), whereas the charge-transfer (CT) component in HLCT continues to maintain the high sensitivity of fluorophores. Under high pressure, pCN-TPA exhibits a large-scale red shift from 509 nm to 608 nm. Ma et al. focused on the changes produced by grinding; thus, such topic will not be discussed in detail here. However, their research might play an important role in future research.
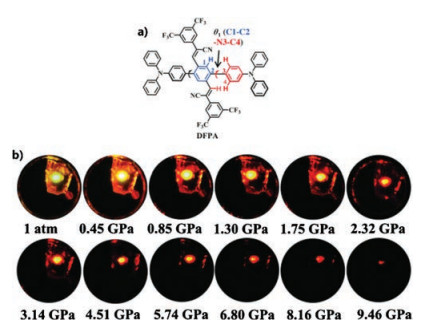
Zhang et al. [55] reported a D-A type cyano-substituted oligo(pphenylenevinylene) (cyano-OPV) derivative (mF-TPA) with TPA (as donor) and DiCN (as acceptor) that reveals a behavior similar to that of the HLCT state with high emission efficiency of up to 82.6% in the crystalline state. Under high pressure, the mF-TPA crystal redshift range was up to 146 nm. Moreover, the fluorescence spectra returned to its original state after pressure release. A series of studies have shown that under high pressure, the molecules are more densely packed, and the interaction between molecules is enhanced. Closer packing and shorter intermolecular distances help increase the polarization effect of adjacent molecules. Eventually, the S1 energy level and the radiative rate constant are gradually reduced, resulting in a red shift in the emission wavelength and a decrease in emission intensity. Xu et al. [56] concluded that the ICT state appears to be under high pressure and is more sensitive to external pressure by studying 4-(anthracen-9- yl)-N, N-diphenylaniline. Soon afterwards, Zhang et al. [57] researched a new cruciform-shaped fluorophore (DFPA, shown in Fig. 3a). Under high pressure, DFPA emission color gradually redshifts from orange to deep red with decreased brightness, as shown in Fig. 3b. TPA groups on the horizontal axis provide a large amount of space for molecular rotation, which is beneficial for the formation of the twisted intramolecular charge transfer (TICT) state, whereas an X-shaped rigid skeleton with highly twisted moieties (TPA and cyanodistyrylbenzene) contributes to high PLQY and weakens the intermolecular interactions under high pressure. Experiments and calculations show that the rotation of the TPA under high pressure reduces the dihedral angle, resulting in a decrease in the excitation energy of S1. This phenomenon corresponds to a redshift in the TICT bands during pressurizing process; that is, the planarity of the molecular conformation may be one of the causes of the redshift. At the same time, the reduction of the distance between molecules increases the nonradiative vibration process and the intermolecular interactions, which leads to a red shift in the emission and a decrease in emission intensity. Overall, the analysis of many TPA derivatives cannot be separated from the role of ICT because TPA derivatives exhibit a variety of changes and applications through ICT.

|
Download:
|
| Fig. 3. (a) DFPA structure. (b) Image of DFPA under UV light at various pressures (from 1 atm to 9.46 GPa). Reproduced with permission [48]. Copyright 2018, Wiley. | |
{kind=link}
Interestingly, Zhang et al. [58] developed a nanofibrous film (ANs@PVA) by introducing a cyano-OPV chromophore into poly (vinyl alcohol) (PVA) through an electrospinning process, as shown in Figs. 4a–d. Before this, Yan et al. conducted a relevant research [59]. However, their study is the first example of radiometric piezochromic luminescent properties in an electrospun film and is the first to demonstrate its attractiveness as a pressure sensor. In this letter, the cyano-OPV guest (oMe-TPA) was studied for its piezochromic characteristics. As shown in Fig. 4e, the transparent crystals emit a bright sky-blue fluorescence under ambient pressure. When the hydrostatic pressure increases from 0 GPa to 13 GPa, the PL color of the crystal exhibits a continuous and clear alteration from sky-blue to green, and finally to dark red; the corresponding PL bands reveal a large-scale redshift from 468 nm to 626 nm. What is more, the fitting PL bands revealed a linear relationship with hydrostatic pressure, as shown in Fig. 4f. Upon fully releasing the pressure, all bands return to their original state, indicating excellent stability and reversibility. According to their analysis, the shortened intermolecular distance caused by high pressure favors a polarization effect on neighboring molecules and conformational planarization. As a result, the energy of the lowest excited state and radiative transition rate are reduced, finally leading to the red shift and weakening of PL spectra.

|
Download:
|
| Fig. 4. (a) Schematic illustration of the incorporation of oMe-TPA (b) molecules into PVA fiber as PCL films (c) 2%ANs@PVA) by an electrospinning method. (d) Corresponding SEM images of 2%ANs@PVAfilm. (e) Images of oMe-TPA crystal recorded under UV light (λex = 365 nm) at various pressures (from 1 atm to 13.4 GPa). (f) PL spectra of oMe-TPA crystal under different pressures. Reproduced with permission [58]. Copyright 2018, Wiley. | |
{kind=link}
As an important structural unit of organic luminescent materials, changes in the conformation of TPA under high pressure may affect the conformation of the entire molecule (e.g., molecular rotation, overall electronic structure).
2.3. Boron-containing organic compoundsLumiscent groups, such as TPA, anthracene and TPE, are common. Zhang et al. [60] found another way to discover a new boron-containing material, boron diketonate. In their study, two crystal forms (1OC and 1RC) were obtained and an in-depth analysis was conducted using 1OC as an example. Crystal 1OC holds the triclinic crystal system and P21/c space group, and the unit cell contains one molecule. The boron atom adopts a typical four-coordinate geometry, and the bond lengths and angles are comparable with those of other similar boron compounds. The structure makes 1OC susceptible to tensile forces, as well as to anisotropic and isotropic pressures. Under high pressure, 1OC red shifts (from 580 nm to 660 nm, shown in Fig. 5). They believed that the current luminescent chromism is due largely to intermolecular distance alternations between two molecules within one dimer. From the crystal structural analysis, the distance between two molecules within one dimer is approximately 3.5 Å, which indicates that the dimer has a moderate π–π interaction. Compression may shorten the distance between π planes in the dimer and hence lead to red shifting. Then according to the previous research, they synthesized a new boron-containing compound [61] by refluxing the mixture of 1, 3-diaryl-diketone and fluorobis(pentafluorophenyl)borane in THF. One of the crystals undergoes a significant color shift from orange (580 nm) to red (655 nm, shown in Fig. 5), which can be recovered after pressure relief. Combined with Raman spectroscopy, red shift is found to be caused by a decrease in interatomic distance and an increase in effective force constant. The increase or decrease in the relative intensity can be attributed to the pressure-induced molecular conformation alterations and ligand distortion.

|
Download:
|
| Fig. 5. (a) Boron diketonate crystals under different isotropic pressures. (b) The PL spectra of boron diketonate crystals under different pressures. (c) PL spectra of new boron-containing compound under different pressures. (a, b) Reproduced with permission [60]. Copyright 2015, Wiley. (c) Reproduced with permission [61]. Copyright 2015, The Royal Society of Chemistry. | |
{kind=link}
The above research results show that boron-containing organic compounds are also important piezochromic materials, but designing appropriate molecular structures is necessary. From Zhang's studies, pressure can more precisely regulate the position of the luminescence peak, which has potential applications in pressure sensors.
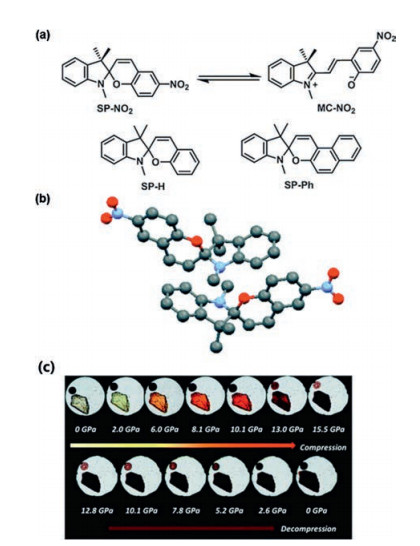
2.4. Spiropyrans (SP) derivativesBenzo[1,3]oxazine (OX-1), the first pressure-controllable molecular switch, was studied by Li et al. [62]. OX-1 has a similar structure to SP, which realizes the bistable state to continuous state transition in a crystal by pressure-enhanced intermolecular interactions. SP is one of the most widely studied thermochromic, solvatochromic and photochromic compounds. It is widely used in functional materials, such as in the improvement of photoelectric performance. Under pressure, SP transforms from a colorless or weakly colored closed form to a highly colored open planar merocyanine (MC) form. Ma et al. [63] used SP-NO2 as an example to study the mechanochromic response of SP via pressure-induced isomerization. Under high pressure, the C–O bond cleavage during the isomerization process leads to the hybridization of the spiro carbon from sp3 to sp2, which is beneficial for the change in the crystal structure from spiral to planar structure. The mechanochromism of SP might also be influenced by an effective charge transfer process from the lone pair of the indoline nitrogen into the antibonding orbital of the C–O spiro bond. The experimental phenomenon also showed that under high pressure, the crystal color changed from light yellow to black red, indicating that transition from SP-NO2 to MC–NO2 occurred (Fig. 6).

|
Download:
|
| Fig. 6. (a) Isomerization and structures of spiropyrans. (b) ORTEP drawings of the crystal structures of SP-NO2. (c) In situ micrographs of SP-NO2 crystal under 0–15.5 and 0 GPa after the discharge of pressure. Adapted with permission [63]. Copyright 2015, The Royal Society of Chemistry. | |
{kind=link}
Also based on SP, Ma et al. [64] selected pyrene as a chromophore to study the multicolor switching of the spiropyran derivative (SP-Pyr). At high pressure (up to 8.3 GPa), the emission peaks are constantly red shifted from 424 nm to 494 nm. When the pressure reaches 15 GPa and is released, a new emission peak (671 nm) appears, and one part of the emission of pyrene moieties is reverted to its initial state (424 nm), whereas the other part is shifted to 467 nm as a result of the irreversible changes in pyrene stacking states after the isomerization reaction. Ground samples exhibit a color change from blue to red. The multimechanochromic responses of SP-Pyr to anisotropic grinding/ shearing and isotropic compression are realized as a result of the combination of the supramolecular structural changes in pyrene moieties and the chemical structural breakage of spiropyrans upon mechanical disturbance. A wide stimuli region is observed to be attributed to a rigid linker that bridges the two moieties, resulting in colors nearly covering the entire visible region. Given the distinct strengths of covalent and noncovalent interactions, the multi-mechanochromic properties can be modulated by the strength of the external force. Furthermore, the multicolored responses can also be distinct by using different modes of stimuli, namely, anisotropic grinding and isotropic compression. In the anisotropic grinding mode, the transformation from pyrene excimers with partially overlapped stacking to sandwich-like packing excimers and further to the ring-opening state of spiropyran accounts for the tricolored switching luminescence. For the isotropic compression, the conformational changes or enhanced excimers and isomerization reaction contribute to the continuous mechanochromic switch.
2.5. Research on other piezochromic systemsCombining the analysis of boron-containing compounds with the above-described anthracene derivatives and TPA derivatives, the effect of high pressure on luminescence affects the crystal structure. Changes in the crystal structure lead to changes in the distance between molecules, intermolecular interactions, and so on. At present, most of the research on piezochromic luminescent materials are scattered, such as those by Yamaguchi, Lu and Yan. However, these studies have laid a solid foundation for the study of luminescence under high pressure.
Yamaguchi et al. [65] conducted a research on tetrathiazolylthiophene. This compound exhibits a remarkable change: when it is mechanically ground, it produces a blue shift, whereas when the pressure is applied using DAC, a red shift occurs. The sample exhibits a 556 nm emission before grinding, and after grinding, it emits a large blue shift to 490 nm. When the crystal is ground, the degree of the intermolecular hydrogen bond is disturbed, the weak 3D hydrogen bond network is in a disordered state, and another long-range ordered phase is not formed. Correspondingly, the face-to-face dimer structure may be deformed into a disordered structure. As a result, the yellow-emitting excimer is no longer formed; instead, the distance between the neighboring fluorophores changes, resulting in green emission. In Fig. 7, during the compression process, the emission peak is red shifted from 556 nm to 609 nm (3.2 GPa), and the emission intensity is gradually weakened. When the pressure is gradually returned to ambient pressure, the emission peak returns to 556 nm. The test results show that the hydrogen bond lattice is deformed, which reduces the void space between the face-to-face 2, 5-dithiazolylthiophene moieties. The two teraryl skeletons slide along the long axis such that the units overlap each other to a greater extent, forming a dimer. This phenomenon may be the reason for the red shift emission under hydrostatic pressure. Moreover, due to the strengthening of hydrogen bonds, the emission is weakened.

|
Download:
|
| Fig. 7. Fluorescence microspectroscopy of one (crystal) under high pressure: (a) compression and (b) decompression processes in the range of 0.1–3.2 GPa. (c) Micrographs of the crystal under high pressure. A UV LED lamp was used for the excitation (λex = 365 nm). Reprinted with permission [65]. Copyright 2013. American Chemical Society. | |
{kind=link}
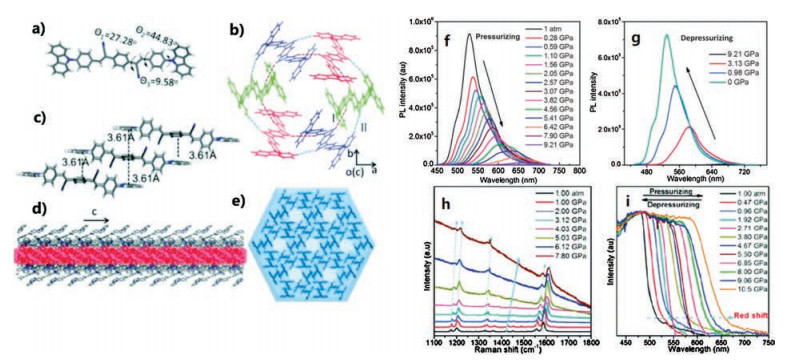
Lu et al. [66], through a classical Knoevenagel reaction between carbazolebenzaldehyde and p-phenylenediacetonitrile, synthesized (2z, 2'z)-2, 2'-(1, 4-phenylene)bis(3-(4-(9H-carbazol9-yl)phenyl)acrylonitrile (CzCNDSB) (Figs. 8a–e), which is a typical AIE material. The CzCNDSB crystal adopts a porous, distorted conformation with a cavernous channel, which provides available space to withstand higher external pressures. During the compression process, the volume of the crystal lattice can shrink greatly and lead to large deformations. From 1 atm to 9.21 GPa, crystal emission red shifted from 529 nm to 684 nm. Upon fully releasing the pressure to 1 atm, the emission spectra can completely recover to the original position (Figs. 8f and g). According to in situ UV–vis absorption spectra and Raman spectra of CzCNDSB (Figs. 8h and i), with increased pressure, the decreased interatomic distance leads to the decreased energy gap and the internal and external modes shifted to high frequencies. The decrease in atomic distances generally results in a blue shift of the peak in the internal mode. Moreover, the shortened intermolecular distance can enhance the polarization effect of adjacent molecules, which reduces the energy of the lowest excited state and the radiative transition rate, leading to a redshift in the fluorescent wavelength and a decrease in PL intensity.

|
Download:
|
| Fig. 8. (a) Molecular conformation of CzCNDSB; (b) C-H⋯N hydrogen bonds I and II between adjacent molecules; (c) π–π interaction between molecules along the c axis; (d) Side view of the cavernous channel in the crystal; (e) Top view of the crystal structure. (f) PL spectra of a CzCNDSB crystal during the pressurizing process; (g) PL spectra of the CzCNDSB crystal during the depressurizing process; (h) In situ Raman spectra and (i) in situ absorption spectra of the CzCNDSB crystal during the pressurizing process. Adapted with permission [66]. Copyright 2016, The Royal Society of Chemistry. | |
{kind=link}
Among the many organic luminescent materials, researchers should also study co-crystals, which belongs to the category of supramolecules. Different combinations of co-crystals can produce unexpected results [67-70]. Yan et al. [71] selected 4-bis(1-cyano2- phenyl-ethenyl)benzene (DCSB, A) as the core chromophore to self-assemble with two small organic molecules, tetrafluorohydroquinone (B) and 2, 3, 5, 6-tetrafluoro-4-hydroxy-benzoic (C), to study their relationship with solid-state molecular arrangement and piezochromic luminescence. They obtained two new cocrystals, A-5B and A-2C, which are organized via multiple hydrogen bonds and π–π stacking interactions. Given the differences in the molecular structures of B and C, the two assembled structures exhibit significant differences in supramolecular interactions. The first is the emission under ambient pressure. A-2C exhibits a sky-blue luminescence at 458 nm, which is close to the emission of A crystal. The A-5B exhibits a green emission at 498 nm (Figs. 9α–γ). When pressure is applied to the two cocrystals by DAC, they produce similar results with a significant red shift in the emission peak and a partial recovery of the emission intensity as the pressure is reduced (pressure relief), as shown in Figs. 9a–f. This phenomenon is related to the collapse and reconstruction of supramolecular structures. When the co-crystals are subjected to an external force, the molecules spontaneously change their positions and conformations to accommodate the volume reduction caused by the compression process, leading to the fracture of the weak hydrogen bond and the enhancement of intermolecular interaction, eventually resulting in fluorescence quenching and red shift. After depressurization, supramolecular interactions are reconstructed. The luminescence is partially restored. In this study, by introducing suitable co-forms, the piezochromic materials of AIE and two-component molecular materials can be highly regulated due to the alternation of the intermolecular interactions, stacking manner, and molecular conformation. This work provides new ideas for the development of new AIE materials and piezochromic materials from a supramolecular point of view.

|
Download:
|
| Fig. 9. Fluorescence images and dihedral angles between intermediate benzene ring and bilateral benzene rings of (α) DCSB (A) and DCSB-based co-crystals: (β) A- 2C; (γ) A-5B. In situ PL spectra of co-crystals A-2C (a–c) and A-5B (d–f) under high pressure from 1 atm to 10 GPa. Adapted with permission [71]. Copyright 2018, The Royal Society of Chemistry. | |
{kind=link}
The development of DAC provides an ideal research tool for studying piezochromic materials. This section summarizes various luminescent systems, such as anthracene, TPA, boron-containing compounds, and organic compounds. We also summed up some rules: under ambient pressure, the emission of samples is weakened and red shifted. The change in the material can be attributed to the following main points: (1) Change in pressure, which causes the change in molecular aggregation and the planarization of the molecule; (2) Decrease in intermolecular distance and influence of different angles of molecular rotation; (3) Change in atomic hybridization, such as sp3→ sp2; (4) ring-opening reaction of the molecule containing benzene ring.
When returning from high pressure to ambient pressure, if the material does not undergo structural damage, ring-opening reaction, etc., the emission can be restored to the initial state. These materials produce novel piezochromic behavior under high pressure. Of course, some materials will blueshift under high pressure, but few related studies have reported such materials. Typically, these materials lead to molecular planarization or form a more closely packed structure under isotropic compression of DAC, causing a red shift. On this basis, some luminescent materials with loose structures may be good candidates for achieving piezochromic behavior.
3. Pressure-induced emission enhanced materialsNormally, as the pressure increases, the fluorescence emission gradually decreases. However, some materials exhibit a different behavior during this phenomenon; the emission intensity of such materials increases with increased pressure. This phenomenon is called pressure-induced enhanced emission (PIEE). Some materials exhibit no emission under ambient pressure but demonstrate emission under high pressure; this phenomenon is called pressure-induced emission (PIE). At present, the proposed mechanical model for explaining the main causes of the aggregation induced emission (AIE) phenomenon is the restriction of intramolecular motion (RIM), which involves the restriction of intramolecular vibrations (RIV) and the restriction of intramolecular rotations (RIR) [72-75]. The effect of high pressure on vibration and rotational motion is due to the fact that pressure can reduce the intermolecular distance [76]. Many PIE/PIEE materials demonstrate a similar mechanism to AIE materials. This finding was also demonstrated by Shuai et al. [77] in the theoretical calculation of the hexaphenylsilole PIE experiment. In this part, we discuss PIEE materials.
3.1. Tetraphenylethene and its derivativesTetraphenylethene (TPE) is a prototype AIE-active chromophore with highly twisted conformations locked by multiple aromatic C– H⋯π and C–H⋯C contacts. Based on its structure, this prototype has a butterfly configuration: four benzene rings, which are like the wings of a butterfly that can vibrate and even rotate around the C— C bond between the vinyl and benzene rings. Therefore, its structure and intermolecular interaction can easily be modulated by external pressure to produce a new phenomenon, especially PL. To this end, Zou et al. [35] conducted an in-depth study of TPE under high pressure. With increased pressure on TPE, the PL changed from deep blue (448 nm, 1 atm) to sky blue (467 nm, 5.3 GPa) and finally to spring green (10 GPa, 488 nm), as shown in Fig. 10. Surprisingly, the emission intensity is significantly enhanced in the pressure range of 1.5–5.3 GPa, and the emission intensity at 5.3 GPa is approximately three times that at 1.5 GPa. After depressurization, the PL returns to the original emission band. Combined with high-pressure synchrotron powder XRD experiments, the fluorescence decay profile analysis and IR show that pressure-induced unit cell contraction in TPE is dominated by a significant decrease in intermolecular distance rather than conformational planarization, and hydrogen bonds are formed around 1.44 GPa. Closer intermolecular interactions allow for additional nonradiative decay channels under pressure, which result in the decrease in emission intensity and lifetime of compressed TPE in 0.1 GPa to 1.5 GPa. With increased pressure (1.5–5.3 GPa), the RIR hypothesis for the mechanism of AIE phenomenon in propeller-shaped luminogens becomes true. In other words, the intermolecular interactions lead to the restriction of the aromatic-part movement, which reduces the energy loss through the nonradiative rotational relaxation channel and thus enhances PL efficiency. Moreover, the formation of hydrogen bonds further limits the movement of the aromatic moiety, resulting in PIEE. When the pressure exceeds 5.3 GPa, the TPE exhibits amorphization, and the intermolecular force in the amorphous phase is not as effective as limiting the intramolecular rotation in the crystal, causing a decrease in PL efficiency. Wang et al. [36] also studied triphenylethylene (TriPE, shown in Fig. 11). TriPE has a similar structure to TPE and exhibits a phenomenon similar to that exhibited by PIEE, such as the interpretation of RIM and the formation of related hydrogen bonds. However, a difference is always observed in the fact that TriPE has one less benzene ring than TPE. In terms of phenomenon, the pressure range and pressure of intensity on PIEE are lower than that of TPE. Given the ring-opening reaction of benzene ring in TriPE under high pressure, TPE luminescence can be restored after depressurization and TriPE cannot be restored.

|
Download:
|
| Fig. 10. (a) Pressure-dependent PL maximum (left axis) and intensity (right axis) of TPE crystals. (b) Corresponding typical photographs under UV irradiation (λex = 375 nm). Reprinted with permission [35]. Copyright 2014, American Chemical Society. | |
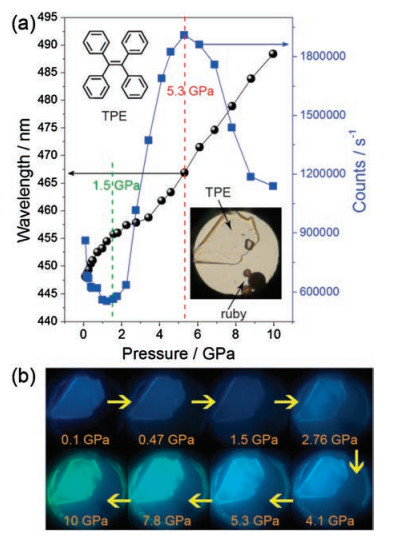{kind=link}

|
Download:
|
| Fig. 11. (a) PL spectra of a TriPE in the pressures range of 0.0–20.4 GPa. Change in the PL intensity represented by red arrows. (b) Corresponding PL photographs at different pressures. (c) Corresponding International Commission on Illumination chromaticity diagram of TriPE under different pressures. The inset of the panel contains the molecular structure of TriPE. Reprinted with permission [36]. Copyright 2019, American Chemical Society. | |
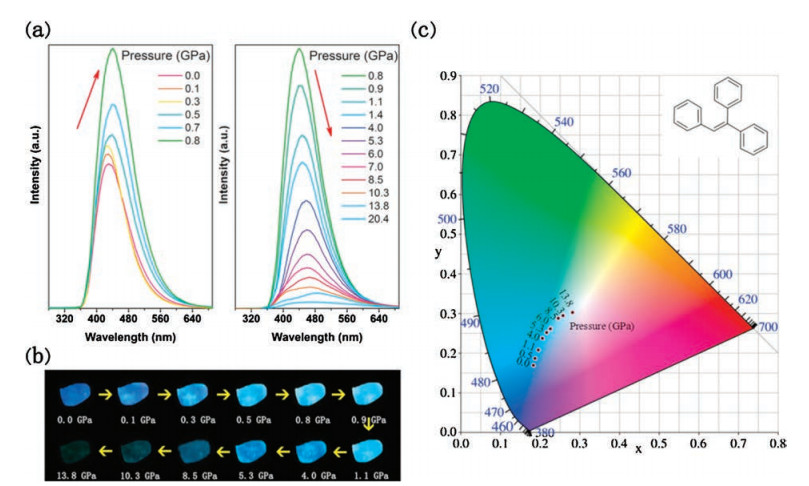{kind=link}
Tian et al. [37] studied the PIEE phenomenon of acridoyltetraphenylethene (AD-TPE). As shown in Figs. 12a and c, AD-TPE is a D-A molecule incorporating a twisted TPE unit as an electron donor and a rigid AD unit as an electron acceptor. Referring to the D-A molecule, the ICT process was mentioned in the section on TPA derivative. In the structure of the AD-TPE, the nearly orthogonal conformation between TPE and AD completely separates the electron distribution and inhibits the ICT process, resulting inLE state emission in the non-luminescent phase of the molecular crystal. When the molecule is under mechanical stimulation, the twisted conformation can be altered by force perturbation, resulting in overlapping frontier orbitals between the donor and the acceptor and the formation of ICT state. Thus, the switching of excited state characteristics by the mechanical stimuliinduced the change in luminescence from the nonemission nonluminous-phase to the bright cyan emission luminescence-phase (Figs. 12b and d).
TPE plays an important role in pressure-induced emission, and the butterfly structure gives TPE a rich variation (adding different groups to the benzene ring). The presentation and development of the RIM model paved the way for the study of TPE.

|
Download:
|
| Fig. 12. (a, b) Visible and fluorescence images of a single crystal of AD-TPE under different hydrostatic pressures. (c) Structure of AD-TPE. (d) Corresponding fluorescence spectra. Reproduced with permission [37]. Copyright 2015, Wiley. | |
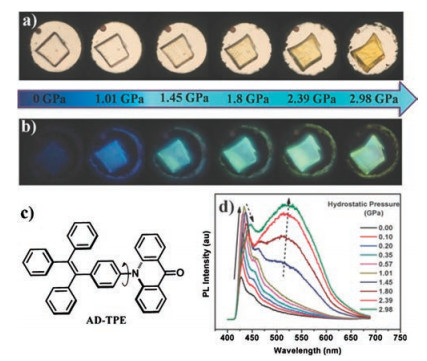{kind=link}
3.2. Triphenylamine (TPA) derivatives
In the piezochromic section, some TPA derivatives under high pressure were studied. Given that TPA exhibits a PIEE phenomenon, it will again be described here. Xu et al. [38] conducted a study on TPA under high pressure. Under pressure, TPA emission intensity increases several times (Fig. 13), and a weak red shift occurs. According to related research, TPA does not undergo phase transition under high pressure but forms a new metastable structure. Although the molecular spacing is reduced, no π–π stacking is observed, thereby producing a weak red shift. At the same time, given that the rotation and vibration of the benzene ring are limited (RIR and RIV), the nonradiative transition is attenuated, resulting in enhanced fluorescence. Zhang et al. [39] reported that (Z)-3-(4'-(diphenylamino)-[1, 1'-biphenyl]-4-yl)-2- (4-methoxyphenyl)-acrylonitrile (β-CN-TPA) shows PIEE phenomenon under high pressure. Below 1.07 GPa, the emission of β-CNTPA exhibits a weak red shift and significant fluorescence enhancement. When the pressure is between 1.07 GPa and 9.97 GPa, a large-scale red shift occurs, and the emission intensity is rapidly weakened. They also explained the PIE phenomenon based on the RIR. However, a π–π interaction occurs between molecules in β-CN-TPA and strengthens with increased pressure, resulting in a large-scale red shift.

|
Download:
|
| Fig. 13. Fluorescence spectra of TPA under different pressures via DAC. The excitation wavelength was 365 nm. (Inset) Pressure versus peak wavelength. Reprinted with permission [38]. Copyright 2015, American Chemical Society. | |
{kind=link}
Uniquely, 4-(2-(4'-(diphenylamino)-[1, 1'-biphenyl]-4-yl)-1Hphenanthro[9, 10-d]imidazol-1 yl)benzonitrile (TBPMCN, Fig. 14a) was investigated by Yang et al. [40] Their study showed that a unique rehybridization of nitrogen atom induces PL enhancement under pressure stimulation. When the pressure is in the range of 0– 2.04 GPa and 4.55–13.2 GPa, the emission intensity of the crystal decrease with increased pressure. Abnormally, when the pressure increases from 2.50 GPa to 4.55 GPa, the emission intensity of the crystal increases significantly. The emission peak is red shifted continuously (Figs. 14b and c). Through the analysis of its structure, several stages (Fig. 14d) can be clearly identified by increasing external pressure: At lower pressures (0–2.04 GPa), the adjacent molecules will be squeezed by the enhanced π–π interaction, which results in a decrease in emission intensity. When the pressure is further enhanced (2.50–4.55 GPa), the energy barrier from the triangular-cone (TC) to the three-blade-propeller (TBP) conformation transition can be overcome, and the N atom is rehybridized from sp3 to sp2. Thus, the TPA moiety with the TC conformation is compressed into a thermodynamically stable TBP conformation through the C— H— N hydrogen bonding. The TBP conformation is more suitable for efficient emission due to the formation of the HLCT emissive state, which essentially exhibits the PIE phenomenon at this stage. When the pressure is high enough (4.55–13.2 GPa), the TBPMCN molecule exhibits a decrease in emission intensity, a red shift of the emission peak and a broadening of the emission peak. This result can be attributed to the intermolecular formation of new excited state species with lower energy and lower emission, as a result of the increasing π–π interaction and the CT-dominated excited state property in the solid state of TBPMCN.
The TPA derivatives belong to a huge family. Similar to TPE, RIM is an important theory for explaining the PIE phenomenon of TPA derivatives. TPA derivatives cannot be ignored when exploring new materials with PIE phenomenon.
3.3. Research on other PIEE systemsIn addition to the TPE and TPA and their derivatives studied by many researchers, many compounds still exhibit the PIEE phenomenon, such as Yang's early work on PIEE [41, 76]. Some people have discovered new chromophores, whereas others have synthesized new materials from different angles; however, no systematic research has been conducted. These works play an important role in the exploration of new PIE materials. Here, carbazole and a new co-crystal will be described.
Zou et al. [42] conducted a research on carbazole, which is a typical blue-emitting organic fluorophore. The crystal structure is formed by alternating layers along the c-axis. Within each layer, molecules are linked by intermolecular N— H⋯π hydrogen bonds and C— H⋯π hydrogen bonds, exhibiting herringbone motifs. Below 1.0 GPa, the carbazole emission intensity increases with increased pressure. However, when the pressure exceeds1 GPa, emission intensity decreases with increased pressure (Fig. 15). According to its structure and experiments, this phenomenon is attributed to the increased N— H⋯π interaction as the pressure value of lower than 1 GPa increases. The N— H⋯π interaction is limited to the N— H stretching vibration, thereby suppressing the nonradiative vibration process to cause emission enhancement. However, excessive pressure causes the distance between the planes of the crystal molecules to decrease, which promotes an effective π–π interaction, resulting in emission reduction and red shift. The carbazole research provides direct experimental evidence for RIV and provides a new foundation for the study of other potential applications of PIEE materials.

|
Download:
|
| Fig. 14. (a) Chemical structure of TBPMCN molecule. Piezochromic PL spectra of TBPMCN crystal (b) and amorphous powder (c). (d) Assumed mechanism diagram of RHIEE in TBPMCN crystal. Reproduced with permission [40]. Copyright 2017, Wiley. | |
{kind=link}

|
Download:
|
| Fig. 15. (a) PL spectra of a carbazole crystal in range of pressures from 0.0 GPa to 10.3 GPa excited by a 355 nm laser. The red arrows represent the changes in the PL intensity. (b) Corresponding PL photographs under high pressure. Reprinted with permission [42]. Copyright 2017, American Chemical Society. | |
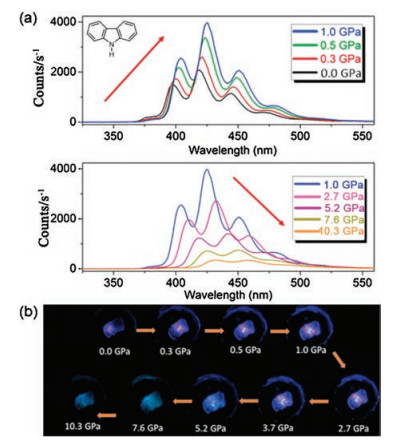{kind=link}
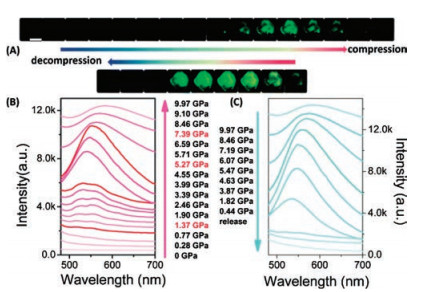
In the previous section, the research on co-crystal piezochromic materials is mentioned. Such materials produce various wonderful phenomena based on supramolecular interactions. Given that cocrystal has a variety of self-assembly methods, this feature greatly expands the new organic luminescent materials. Cui et al. [43] designed a series of two-component co-crystals driven by I⋯N interactions based on the bipyridine (BIPY) chromophore with one among three different co-former building blocks, iodopentafluorobenzene (IPFB), 1, 4-diiodotetrafluorobenzene (DITFB) and 1, 3, 5- trifluoro-2, 4, 6-triiodobenzene (IFB).The three co-crystals are assembled via the designed halogen bonds between the pyridine group (N atom) in BIPY and the halogen units (C–I) in the coformers. A series of research were performed with BIPY-DITFB as the representative. As shown in Fig. 16, the BIPY-DITFB emission intensity is continuously enhanced with increased pressure and then decreases. With increased pressure, the I⋯N distance between BIPY and DITFB gradually decreases. Theoretical calculations confirm this conjecture. Theoretical calculations and corresponding experiments show that the pressure can effectively reduce the distance of I⋯N, which is accompanied by the formation of a new CT state, and the pressure can increase the angle of C–I⋯N and enhance the interaction of I⋯N, resulting in an increase in fluorescence efficiency, showing a PIEE phenomenon. However, when the pressure exceeds 7 GPa, the distance between adjacent BIPYor DITFB molecules is gradually shortened. The angle of C–I⋯N decreases, which may slightly weaken the interaction of I⋯N, resulting in low fluorescence efficiency.
Currently, research on pressure-induced emission is still in its infancy. Whether it is TPE, TPA, or carbazole, it is explained by RIM as the main theory. This provides a structural reference and orientation for exploring new PIEE materials. However, Cui et al. studied new PIEE materials from a supramolecular perspective, which enriches potential candidates for PIEE materials. Perhaps more types of PIEE materials are yet to be discovered.

|
Download:
|
| Fig. 16. (A) Photos of the BIPY–DITFB co-crystal under different pressure values. The scale bar: 100 μm. The fluorescence spectra of the BIPY–DITFB co-crystal during compression (B) and decompression (C) via DAC. Excitation wavelength was 365 nm. Reproduced with permission [43]. Copyright 2018, The Royal Society of Chemistry. | |
{kind=link}
4. Summary
In this review, the latest research progress on piezochromic materials and pressure-induced emission materials are summarized, and the material structure, luminescence properties, and mechanism are expounded. Compared with inorganic luminescent materials, organic luminescent materials have the advantages of variety, good adjustability, rich color, and flexible molecular design, making them potential materials for large-scale application. Nevertheless, the number of piezochromic materials, especially the pressure-induced emission enhancement materials, is relatively rare. In-depth relevant research must also be conducted, and new varieties must be further developed. At present, the practical application of these two materials remains relatively rare, although these two materials have great potential applications in theory. Hence, future research must be performed on the bases of these practical applications.
AcknowledgmentsThis work is supported by the National Natural Science Foundation of China (NSFC, Nos. 21725304 and 11774120), the Chang Jiang Scholars Program of China (No. T2016051), the Fundamental Research Funds for the Central Universities.
| [1] |
Y.Y. Qin, W.J. Xu, C.Y. Hu, S.J. Liu, Q. Zhao, Chin. J. Inorg. Chem. 33 (2017) 1705-1721. |
| [2] |
S.K. Wu, Prog. Chem. 17 (2005) 15-39. |
| [3] |
Y. Sagara, T. Kato, Nat. Chem. 1 (2009) 605-610. DOI:10.1038/nchem.411 |
| [4] |
G.J. Xiao, Y. Cao, G.Y. Qi, et al., J. Am. Chem. Soc. 139 (2017) 10087-10094. DOI:10.1021/jacs.7b05260 |
| [5] |
G.J. Xiao, X.Y. Yang, X.X. Zhang, et al., J. Am. Chem. Soc. 137 (2015) 10297-10303. DOI:10.1021/jacs.5b05629 |
| [6] |
Q.X. Zeng, K. Wang, B. Zou, J. Am. Chem. Soc. 139 (2017) 15648-15651. DOI:10.1021/jacs.7b10292 |
| [7] |
Q.X. Zeng, K. Wang, Y.C. Qiao, X.D. Li, B. Zou, J. Phys. Chem. Lett. 87 (2017) 1436-1441. |
| [8] |
G.Y. Qi, K. Wang, G.J. Xiao, B. Zou, Sci. China Chem. 61 (2018) 276-280. DOI:10.1007/s11426-017-9152-8 |
| [9] |
S.Y. Lu, G.J. Xiao, L.Z. Sui, et al., Angew. Chem. Int. Ed. 56 (2017) 6187-6191. DOI:10.1002/anie.201700757 |
| [10] |
C. Liu, G.J. Xiao, M.L. Yang, et al., Angew. Chem. Int. Ed. 57 (2018) 1893-1897. DOI:10.1002/anie.201711409 |
| [11] |
Y.J. Liu, Q.X. Zeng, B. Zou, et al., Angew. Chem. Int. Ed. 57 (2018) 15670-15674. DOI:10.1002/anie.201810149 |
| [12] |
Q. Li, Y.G. Wang, W.C. Pan, et al., Angew. Chem. Int. Ed. 56 (2017) 15969-15973. DOI:10.1002/anie.201708684 |
| [13] |
J. Wu, Y.Y. Cheng, J.B. Lan, et al., J. Am. Chem. Soc. 138 (2016) 12803-12812. DOI:10.1021/jacs.6b03890 |
| [14] |
R.J. Hemley, Annu. Rev. Phys. Chem. 51 (2000) 763-800. DOI:10.1146/annurev.physchem.51.1.763 |
| [15] |
P.F. McMillan, Chem. Soc. Rev. 35 (2006) 855-857. DOI:10.1039/b610410j |
| [16] |
R. Miletich, D.R. Allan, W.F. Kuhs, Rev. Mineral. Geochem. 41 (2000) 445-519. DOI:10.2138/rmg.2000.41.14 |
| [17] |
S. Klotz, J.C. Chervin, P. Munsch, G. Le Marchand, J. Phys. D: Appl. Phys. 42 (2009)075413.
|
| [18] |
H.K. Mao, J. Xu, P.M. Bell, J. Geophys. Res. 91 (1986) 4673-4676. DOI:10.1029/JB091iB05p04673 |
| [19] |
K. Wang, S.R. Li, X. Tan, et al., Chin. Sci. Bull. 59 (2014) 5258-5268. DOI:10.1007/s11434-014-0615-9 |
| [20] |
X.Q. Zhang, Z.Y. Ma, M.Y. Liu, et al., Tetrahedron 69 (2013) 10552-10557. |
| [21] |
Z.Y. Ma, M.J. Teng, Z.J. Wang, X.R. Jia, Tetrahedron Lett. 54 (2013) 6504-6506. DOI:10.1016/j.tetlet.2013.09.083 |
| [22] |
Y.J. Zhang, K. Wang, G.L. Zhuang, et al., Chem. Eur. J. 21 (2015) 2474-2479. DOI:10.1002/chem.201405348 |
| [23] |
Y. Sagara, K. Kubo, T. Nakamura, N. Tamaoki, C. Weder, Chem. Mater. 29 (2017) 1273-1278. DOI:10.1021/acs.chemmater.6b04720 |
| [24] |
M. Sase, S. Yamaguchi, Y. Sagara, et al., J. Mater. Chem. 21 (2011) 8347-8354. DOI:10.1039/c0jm03950k |
| [25] |
X. Meng, G.Y. Qi, C. Zhang, et al., Chem. Commun. 51 (2015) 9320-9323. DOI:10.1039/C5CC01064K |
| [26] |
Z.W. Ma, Z. Liu, S.Y. Lu, et al., Nat. Comm. 9 (2018) 4506-4513. DOI:10.1038/s41467-018-06840-8 |
| [27] |
Y. Shi, Z.W. Ma, D.L. Zhao, et al., J. Am. Chem. Soc. 141 (2019) 6504-6508. DOI:10.1021/jacs.9b02568 |
| [28] |
L. Zhang, L.W. Wu, K. Wang, B. Zou, Adv. Sci. 6 (2019) 1801628.
|
| [29] |
L. Zhang, C.M. Liu, L.R. Wang, et al., Angew. Chem. Int. Ed. 57 (2018) 11213-11217. DOI:10.1002/anie.201804310 |
| [30] |
L. Zhang, C.M. Liu, Y. Lin, et al., J. Phys. Chem. Lett. 10 (2019) 1676-1683. DOI:10.1021/acs.jpclett.9b00595 |
| [31] |
Y.P. Chen, R.J. Fu, L.R. Wang, et al., J. Mater. Chem. A 7 (2019) 6357-6362. |
| [32] |
G.J. Xiao, Y.N. Wang, D. Han, et al., J. Am. Chem. Soc. 140 (2018) 13970-13975. DOI:10.1021/jacs.8b09416 |
| [33] |
P.F. Lv, S.R. Yang, C. Liu, et al., J. Phys. Chem. C (2019) 15339-15344.
|
| [34] |
Z.A. Dreger, J.O. White, H.G. Drickamer, Chem. Phys. Lett. 290 (1998) 399-404. DOI:10.1016/S0009-2614(98)00580-6 |
| [35] |
H.S. Yuan, K. Wang, K. Yang, B.B. Liu, B. Zou, J. Phys. Chem. Lett. 5 (2014) 2968-2973. DOI:10.1021/jz501371k |
| [36] |
N. Li, Y.R. Gu, Y.P. Chen, et al., J. Phys. Chem. C 123 (2019) 6763-6767. DOI:10.1021/acs.jpcc.9b00670 |
| [37] |
Q.K. Qi, J.Y. Qian, X. Tan, et al., Adv. Funct. Mater. 25 (2015) 4005-4010. DOI:10.1002/adfm.201501224 |
| [38] |
J.X. Wu, H.L. Wang, S.P. Xu, W.Q. Xu, J. Phys. Chem. A 119 (2015) 1303-1308. DOI:10.1021/jp511380a |
| [39] |
M. Ouyang, L.L. Zhan, X.J. Lv, et al., RSC Adv. 6 (2016) 1188-1193. DOI:10.1039/C5RA21218A |
| [40] |
S.T. Zhang, Y.X. Dai, S.Y. Luo, et al., Adv. Funct. Mater. 27 (2017) 1602276.
|
| [41] |
Q. Wang, S.Y. Li, L.M. He, et al., Chem. Phys. Chem. 9 (2008) 1146-1152. DOI:10.1002/cphc.200700837 |
| [42] |
Y.R. Gu, K. Wang, Y.X. Dai, et al., J. Phys. Chem. Lett. 8 (2017) 4191-4196. DOI:10.1021/acs.jpclett.7b01796 |
| [43] |
A.S. Li, J. Wang, Y.J. Liu, et al., Phys. Chem. Chem. Phys. 20 (2018) 30297-30303. DOI:10.1039/C8CP06363J |
| [44] |
B.R. Crenshaw, M. Burnworth, D. Khariwala, et al., Macromolecules 40 (2007) 2400-2408. DOI:10.1021/ma062936j |
| [45] |
Y.J. Zhang, Q.B. Song, K. Wang, et al., J. Mater. Chem. C 3 (2015) 3049-3054. DOI:10.1039/C4TC02826K |
| [46] |
K. Wang, H.Y. Zhang, S.Y. Chen, et al., Adv. Mater. 26 (2014) 6168-6173. DOI:10.1002/adma.201401114 |
| [47] |
X. Cheng, K. Wang, S. Huang, et al., Angew. Chem. Int. Ed. 54 (2015) 8369-8373. DOI:10.1002/anie.201503914 |
| [48] |
B.L. Tang, H.P. Liu, F. Li, Y. Wang, H.Y. Zhang, Chem. Commun. 52 (2016) 6577-6580. DOI:10.1039/C6CC02616H |
| [49] |
L. Wang, K.Q. Ye, H.Y. Zhang, Chin. Chem. Lett. 27 (2016) 1367-1375. DOI:10.1016/j.cclet.2016.06.049 |
| [50] |
B.L. Tang, H.P. Liu, F. Li, Y. Wang, H.Y. Zhang, Chem. Commun. 52 (2016) 6577-6580. DOI:10.1039/C6CC02616H |
| [51] |
Y.J. Dong, B. Xu, J.B. Zhang, et al., Angew. Chem. Int. Ed. 51 (2012) 10782-10785. DOI:10.1002/anie.201204660 |
| [52] |
Y.J. Dong, J.B. Zhang, X. Tan, et al., J. Mater. Chem. C 1 (2013) 7554-7559. DOI:10.1039/c3tc31553c |
| [53] |
B. Shao, R.H. Jin, A.S. Li, et al., J. Mater. Chem. C 7 (2019) 3263-3268. DOI:10.1039/C9TC00051H |
| [54] |
H.C. Liu, Y.X. Dai, Yu Gao, et al., Adv. Optical Mater. 6 (2018) 1800085.
|
| [55] |
Y.J. Zhang, M. Qile, J.W. Sun, et al., J. Mater. Chem. C 4 (2016) 9954-9960. DOI:10.1039/C6TC03157A |
| [56] |
A.S. Li, Z.Y. Ma, J.X. Wu, et al., Adv. Optical Mater. 6 (2018) 1700647.
|
| [57] |
Y.J. Zhang, J.X. Zhang, J.H. Shen, et al., Adv. Optical Mater. 6 (2018) 1800956.
|
| [58] |
H.Y. Yang, Z.H. Sun, C.Y. Lv, et al., Chem. Plus. Chem. 83 (2018) 132-139. |
| [59] |
R. Gao, Y.B. Zhao, X.G. Yang, D.P. Yan, RSC Adv. 5 (2015) 56470-56477. DOI:10.1039/C5RA09130F |
| [60] |
L. Wang, K. Wang, B. Zou, et al., Adv. Mater. 27 (2015) 2918-2922. DOI:10.1002/adma.201500589 |
| [61] |
L. Wang, K. Wang, H.Y. Zhang, et al., Chem. Commun. 51 (2015) 7701-7704. DOI:10.1039/C5CC01113B |
| [62] |
Y. Wang, X. Tan, Y.M. Zhang, et al., J. Am. Chem. Soc. 137 (2015) 931-939. DOI:10.1021/ja511499p |
| [63] |
X. Meng, G. Qi, C. Zhang, et al., Chem. Commun. 51 (2015) 9320-9323. DOI:10.1039/C5CC01064K |
| [64] |
X. Meng, G.Y. Qi, X. Li, et al., J. Mater. Chem. C 4 (2016) 7584-7588. DOI:10.1039/C6TC02578A |
| [65] |
K. Nagura, S. Saito, H. Yusa, et al., J. Am. Chem. Soc. 135 (2013) 10322-10325. DOI:10.1021/ja4055228 |
| [66] |
C.F. Feng, K. Wang, Y.X. Xu, et al., Chem. Commun. 52 (2016) 3836-3839. DOI:10.1039/C5CC09152G |
| [67] |
Y.L. Lu, Y.Q. Tang, H.Y. Lin, et al., Chin. Chem. Lett. 29 (2018) 1541-1543. DOI:10.1016/j.cclet.2017.12.021 |
| [68] |
D.P. Yan, H.J. Yang, Q.Y. Meng, H.Y. Lin, M. Wei, Adv. Funct. Mater. 24 (2014) 587-594. DOI:10.1002/adfm.201302072 |
| [69] |
D.P. Yan, J. Lu, J. Ma, et al., Angew. Chem. Int. Ed. 50 (2011) 7037-7040. DOI:10.1002/anie.201102232 |
| [70] |
D.P. Yan, D.G. Evans, Mater. Horiz. 1 (2014) 46-57. DOI:10.1039/C3MH00023K |
| [71] |
B. Lu, Y.J. Zhang, X.G. Yang, et al., J. Mater. Chem. C 6 (2018) 9660-9666. DOI:10.1039/C8TC02444H |
| [72] |
J. Chen, C.C.W. Law, J.W.Y. Lam, et al., Chem. Mater. 15 (2003) 1535-1546. DOI:10.1021/cm021715z |
| [73] |
Y.N. Hong, J.W.Y. Lam, B.Z. Tang, Chem. Comm. 29 (2009) 4332-4353. |
| [74] |
N.L.C. Leung, N. Xie, W.Z. Yuan, et al., Chem. Eur. J. 20 (2014) 15349-15353. DOI:10.1002/chem.201403811 |
| [75] |
J. Mei, Y.N. Hong, J.W.Y. Lam, et al., Adv. Mater. 26 (2014) 5429-5479. DOI:10.1002/adma.201401356 |
| [76] |
S.Y. Li, Q. Wang, Y. Qian, et al., J. Phys. Chem. A 111 (2007) 11793-11800. DOI:10.1021/jp075301a |
| [77] |
T. Zhang, W. Shi, D. Wang, et al., J. Mater. Chem. C 7 (2019) 1388-1398. DOI:10.1039/C8TC05162C |